Lambda – E. coli co-evolution
Bryan Andrews
How do viral receptors balance efforts to escape predation while maintaining core functions?
Proteins that are exposed to the environment often face the unique evolutionary challenge of acquiring resistance to pathogens that would recognize them while also needing to maintain their core cellular functions. In rich media, bacteria are able to rapidly acquire resistance to cohabiting phages through mutations in the appropriate receptor. This resistance precipitates an evolutionary arms race and frequently leads to the extinction of one species. However, evolution in the wild is expected to follow fluctuating selection dynamics, rather than arms race dynamics, and can support long-term coexistence of hosts and pathogens.
Proteins that are exposed to the environment often face the unique evolutionary challenge of acquiring resistance to pathogens that would recognize them while also needing to maintain their core cellular functions. In rich media, bacteria are able to rapidly acquire resistance to cohabiting phages through mutations in the appropriate receptor. This resistance precipitates an evolutionary arms race and frequently leads to the extinction of one species. However, evolution in the wild is expected to follow fluctuating selection dynamics, rather than arms race dynamics, and can support long-term coexistence of hosts and pathogens.
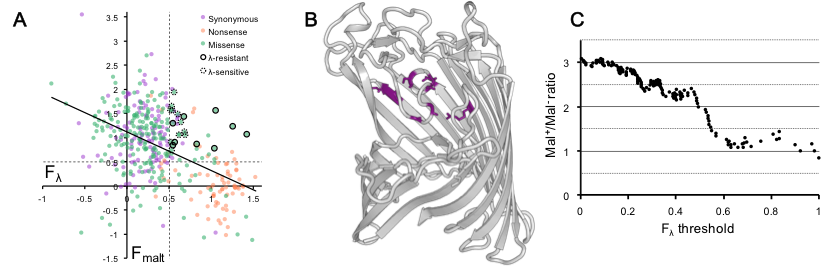
In this project, we assayed thousands of variants of the λ receptor, a maltoporin encoded by the gene lamB, for two traits: resistance to λ and ability to import maltodextrin. By comparing these traits, we can make predictions about how lamB might evolve in the wild. First, we discover that these traits are strongly anti-correlated, implying that most mutations act on a common property, like protein stability, that affects both traits. Second, by focusing on mutations that buck the trend, conferring λ-resistance while maintaining maltodextrin transport, we implicate the region of the receptor that determines binding by λ. Third, by comparing the frequency of λ-resistance that do, or do not, disrupt the native function of lamB, we find that presumption of fluctuating selection dynamics implies near-zero selection on maltose transport in the evolutionary history of lamB.
How do viruses adapt to a dynamic host receptor?
Given that mutations conferring resistance to viruses are abundant, and that many of these resistance mutations are hard to adapt to, one might expect that successful viruses should be a rare occurrence. Instead, viruses outnumber all other biological entities on the planet by an order of magnitude. To resolve this apparent paradox and explain why viruses are so successful, we need a better understanding of what constrains the types of cells a virus can infect, as well as how viruses evolve to overcome resistance and expand their range of potential hosts.
Given that mutations conferring resistance to viruses are abundant, and that many of these resistance mutations are hard to adapt to, one might expect that successful viruses should be a rare occurrence. Instead, viruses outnumber all other biological entities on the planet by an order of magnitude. To resolve this apparent paradox and explain why viruses are so successful, we need a better understanding of what constrains the types of cells a virus can infect, as well as how viruses evolve to overcome resistance and expand their range of potential hosts.
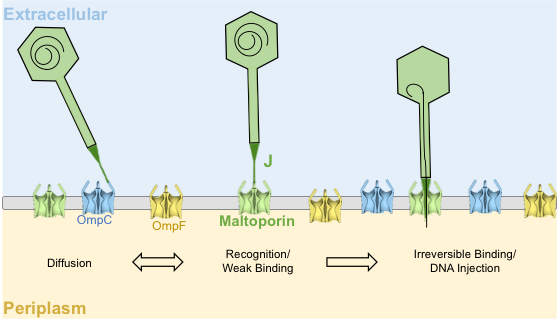
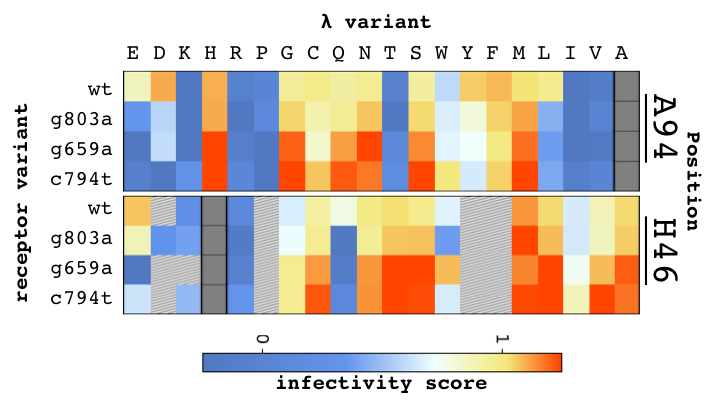
In this project, we are using λ as a model system. Phage λ uses its central tail fiber, the J protein, to infect E. coli via the maltoporin receptor encoded by lamB, and this binding interaction is the primary determinant of whether infection will be successful. We generated a library covering nearly all possible missense mutations in the host range-determining region of J, and assayed these variants for their ability to infect wild type E. coli, generating the heatmap below.
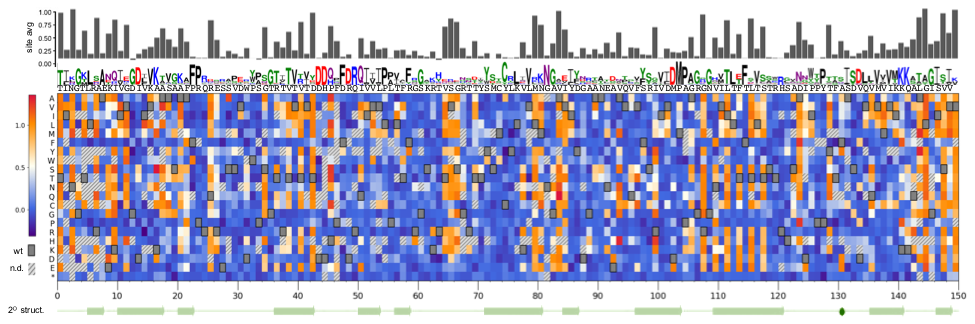
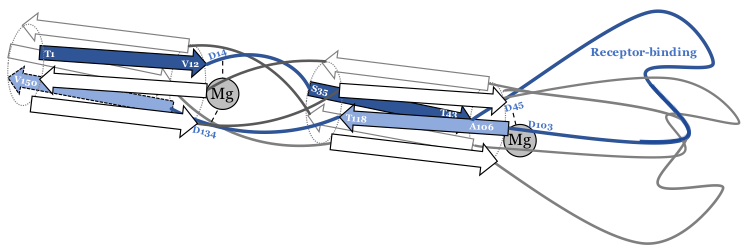
From this analysis, we were able to identify positions where the amino acid identity was important or dispensable, and infer secondary structural elements (namely, beta-sheets). By using this data as well as evolutionary information and published biochemical analyses, we constructed the rough structural model below.
We tested the library of λ variants for the ability to grow on missense variants of lamB that confer resistance to wild type λ. We identified dozens of J mutations that were able to overcome this type of resistance, mostly at positions that evolve rapidly or are known to affect host range. We tested the J library on multiple different missense lamB variants, and we observed many J variants that are enriched on multiple resistant hosts. This observation suggests that there are general mechanisms to overcome resistance by increasing affinity, as opposed to each form of resistance requiring different mutations to overcome it. The abundance of these general counter-resistance mutations may give viruses a leg up when they are challenged to evolve against a host population with multiple distinct resistance mutations.