A covalent protein display for deep mutational scanning of proteins.
Yash Ashok
Deep mutational scanning provides insight into the functions of a protein by measuring the effect of every possible amino acid substitution. The method uses an assay such as growth or fluorescence to select for protein function, and DNA sequencing before and after selection to provide an enrichment score for each variant. However, typical assays do not assess the rich biochemical properties of proteins, such as their thermal stability or kinetics. Here, we couple genotype to biochemical activity by covalently linking a unique RNA barcode to each variant of a protein. The RNA barcodes allow biochemical analyses to be performed on protein variants, with variants in different functional classes identified by sequencing of the barcodes. We attach barcodes using the E. coli tRNA methyl transferase (TrmA) E358Q mutant, which covalently couples to tRNA (Fig. 1). We use FLAG based-affinity enrichment to show that the coupling can occur in E. coli (Fig. 2) and mammalian cells (Fig. 3). We are developing this method to assay protein steady state levels, thermostability and protein interaction.
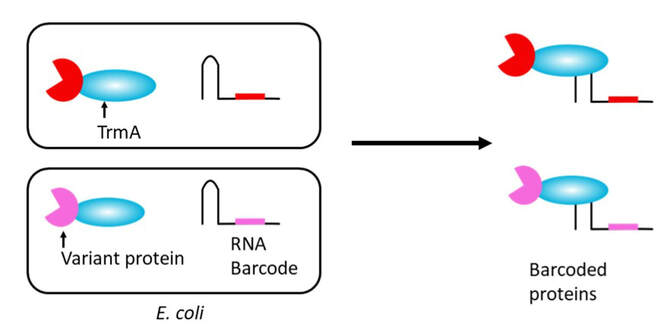
Figure 1: Proposed scheme for barcoding proteins in E. coli. Co-expression of TrmA fusion protein with an RNA barcode leads to production of barcoded proteins.
Terminal protein display (TP-display)
Yash Ashok
The sequencing of human genomes has uncovered many variants in protein-coding regions, but the effects of many of these variants are of unknown significance. These mutations have the potential to modify fundamental properties of proteins, such as their stability, enzymatic activity and affinity for a binding partner. In order to systematically map the effects of protein mutations, we are seeking to develop a new covalent linkage of a protein to its encoding DNA. This phenotype-to-genotype linkage allows us to subject proteins to multiplex biochemical assays and then read out their activities using DNA sequencing.
Some viruses and plasmids employ an unusual type of DNA replication called “protein-primed replication.” In this type of replication, a protein encoded by these viruses and plasmids, which replicate autonomously, serves as a primer, rather than the usual RNA synthesized by primase (Fig. 1A). Φ29 phage, which uses a protein-primed replication, is a model for such replication systems. The Φ29 Terminal protein (TP) is the protein primer that covalently attaches to DNA through a conserved serine. In our approach, we intend to encode a variant library of interest as a fusion protein with TP, which would in turn covalently couple to its own encoding DNA (Fig. 1B).
Some viruses and plasmids employ an unusual type of DNA replication called “protein-primed replication.” In this type of replication, a protein encoded by these viruses and plasmids, which replicate autonomously, serves as a primer, rather than the usual RNA synthesized by primase (Fig. 1A). Φ29 phage, which uses a protein-primed replication, is a model for such replication systems. The Φ29 Terminal protein (TP) is the protein primer that covalently attaches to DNA through a conserved serine. In our approach, we intend to encode a variant library of interest as a fusion protein with TP, which would in turn covalently couple to its own encoding DNA (Fig. 1B).
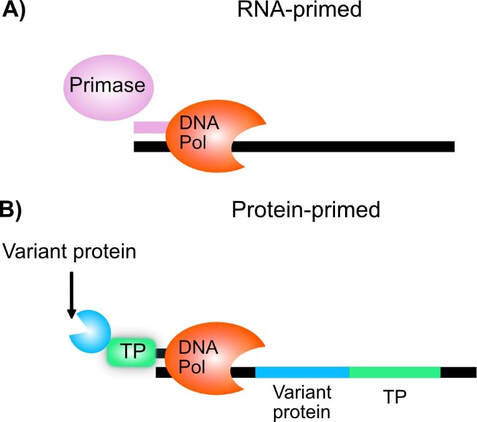
Fig 1: TP-display. A) In RNA-primed replication, primase synthesizes a short RNA primer which is used by DNA polymerases to extend along a template. B) In protein-primed replication Terminal protein (TP) serves as a primer for DNA replication. A variant protein of interest (blue Pacman) is encoded as a fusion protein with TP, which in turn couples to its own encoding DNA during replication, thereby forming a genotype-to-phenotype linkage.
Massively-parallel investigations of inhibitory protein fragments in bacteria
Andrew Savinov
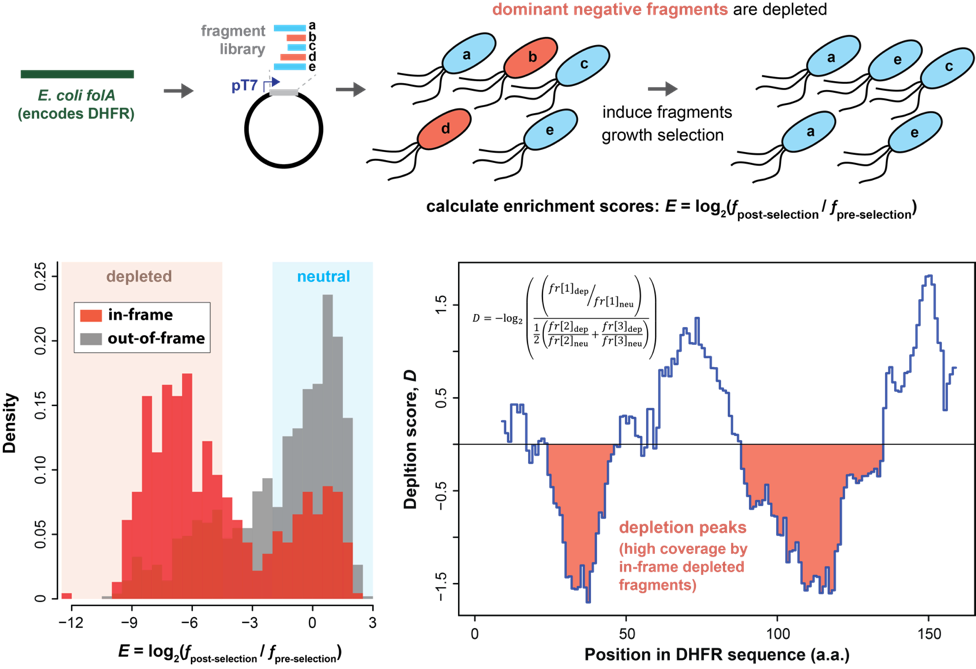
We have adapted an in vivo dominant negative protein fragment screen (Dorrity et al. 2019 Nature Methods 16, 413) for application to bacteria. We construct plasmid libraries containing random polypeptide fragments derived from bacterial genes of interest, expressed under the control of inducible T7 RNAP. We then perform a selection under conditions in which the fragmented genes are important for growth, and use high-throughput sequencing to determine frequencies of fragments before and after selection. Polypeptide fragments that inhibit growth (hence acting as dominant negatives) drop out in the selection. We are using this assay to identify inhibitory fragments of essential bacterial proteins, with the aims of mapping critical functional regions and discovering potent inhibitory peptides that could be developed into novel antibiotics. We are also using this method to study dominant negative inhibitory mechanisms.
In one approach, we are investigating fragments of the well-characterized protein dihydrofolate reductase (DHFR) in E. coli as a model system for these studies. As DHFR is essential in this species, a simple growth selection allows the discovery of dominant negative inhibitors. Given that DHFR is a well-established antibiotic target, the peptide inhibitors we are uncovering could be starting points for novel drug development.
In one approach, we are investigating fragments of the well-characterized protein dihydrofolate reductase (DHFR) in E. coli as a model system for these studies. As DHFR is essential in this species, a simple growth selection allows the discovery of dominant negative inhibitors. Given that DHFR is a well-established antibiotic target, the peptide inhibitors we are uncovering could be starting points for novel drug development.
Genetic tools for assessing the effects of substitutions in protein
Stephanie Zimmerman and Bianca Ruiz (Collaboration with the Villén Lab)
Recent efforts to characterize human genomes have identified millions of protein-altering genetic variants. For most of these variants, the phenotypic effect is unknown. Interpreting variants on this scale will require new, higher-throughput methods to evaluate the effect of mutations on function. In collaboration with the Villén lab in Genome Sciences, we are developing a technology to measure the effect of substitutions across the whole proteome simultaneously.
In this method, substitutions are generated in the entire proteome through errors in translation. Because the variants are not genetically encoded, they cannot be quantified by DNA sequencing, but instead can be detected directly at the protein level by mass spectrometry (MS). In experiments using this technology, we apply a biochemical selection for protein function to a complex pool of protein variants. We use MS to compare the abundance of each protein variant before and after the selection. If a substitution is detrimental for protein function, peptides that contain that substitution will be depleted after selection.
In this method, substitutions are generated in the entire proteome through errors in translation. Because the variants are not genetically encoded, they cannot be quantified by DNA sequencing, but instead can be detected directly at the protein level by mass spectrometry (MS). In experiments using this technology, we apply a biochemical selection for protein function to a complex pool of protein variants. We use MS to compare the abundance of each protein variant before and after the selection. If a substitution is detrimental for protein function, peptides that contain that substitution will be depleted after selection.
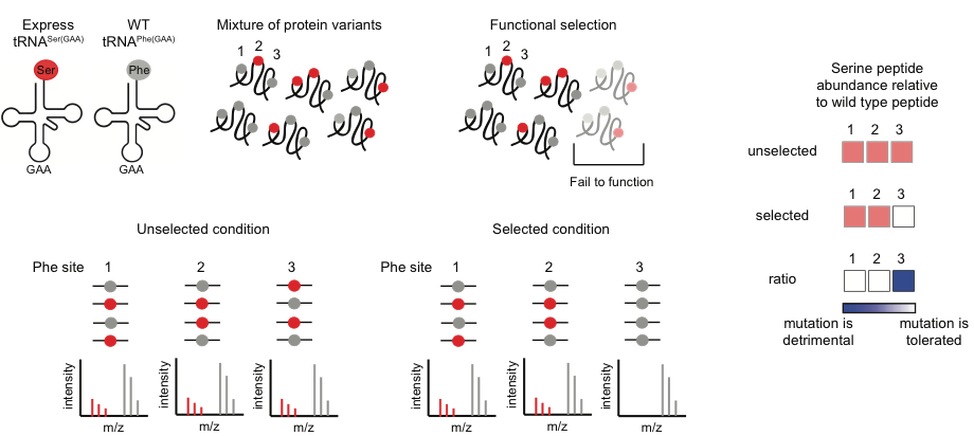
One method to generate mistranslation is to alter tRNA recognition of the codon in mRNA by mutation of the anticodon. As there is a single cytoplasmic amino-acyl tRNA synthetase for each amino acid, tRNAs for amino acids (such as serine and leucine) that are encoded by multiple codons frequently are charged by synthetases that do not use the anticodon for recognition. For example, a serine tRNA with its anticodon of CGA can be mutated to an anticodon of GAA, which binds to a codon specifying phenylalanine. The result of this mutation is the incorporation across the proteome of serines at normally phenylalanine sites. However, altered tRNA recognition can result in levels of mistranslation so high that cells become inviable. For this reason, we are developing inducible systems for mistranslation.
Using a system developed by the Phizicky lab at the University of Rochester, we engineered serine tRNAs that accumulate in cells conditionally. We made all possible anticodon mutants of this serine tRNA and transformed them into yeast in a pool, and identified sequences that cause toxicity when the tRNA accumulates. Using mass
Using a system developed by the Phizicky lab at the University of Rochester, we engineered serine tRNAs that accumulate in cells conditionally. We made all possible anticodon mutants of this serine tRNA and transformed them into yeast in a pool, and identified sequences that cause toxicity when the tRNA accumulates. Using mass
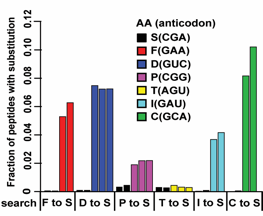
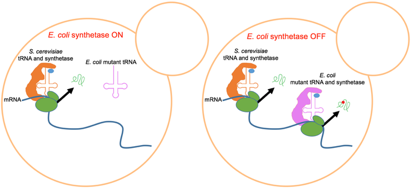
In a second approach, we use conditional expression of a tRNA synthetase. Transcription of mutant tRNAs with altered codon specificity in S. cerevisiae cannot be regulated directly, because eukaryotic tRNA promoter elements are internal to the gene. However, the cognate synthetases, as protein-coding genes, can be placed under the control of an inducible promoter. Furthermore, synthetases and tRNAs derived from a cross-kingdom species (Archaea or bacteria) typically do not interact with yeast synthetases and tRNAs. We harness these characteristics of the translation machinery to generate galactose-inducible expression of an E. coli synthetase along with constitutive expression of its cognate tRNA; this tRNA carries mutations to its anticodon that result in mistranslation. In the absence of galactose, the E. coli tRNA will be expressed in yeast but will be unable to be charged without its synthetase. In the presence of galactose, cells express the E. coli synthetase, resulting in proteome-wide mistranslation.
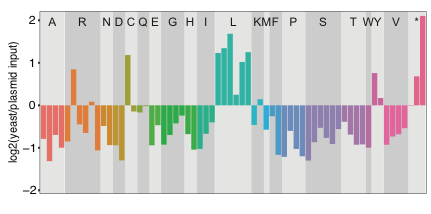
We tested this system using the orthogonal E. coli LeuRS/tRNALeu pair in yeast. We transformed E. coli tRNALeu genes with all possible anticodon sequences in yeast that also express the E. coli LeuRS, grew the cells, and sequenced the population to identify anticodons that cause toxicity. As expected, cells expressing tRNALeu with Leu anticodons enriched in this experiment, suggesting they do not cause toxicity. However, tRNAs with almost all other anticodons depleted, suggesting that they do cause toxicity. These results suggest that E. coli tRNALeu with non-Leu anticodons can produce mistranslation in yeast.
We tested this system using the orthogonal E. coli LeuRS/tRNALeu pair in yeast. We transformed E. coli tRNALeu genes with all possible anticodon sequences in yeast that also express the E. coli LeuRS, grew the cells, and sequenced the population to identify anticodons that cause toxicity. As expected, cells expressing tRNALeu with Leu anticodons enriched in this experiment, suggesting they do not cause toxicity. However, tRNAs with almost all other anticodons depleted, suggesting that they do cause toxicity. These results suggest that E. coli tRNALeu with non-Leu anticodons can produce mistranslation in yeast.
|
|
Identifying variants that alter protein stability in yeast using mistranslation mutagenesis
Stephanie Zimmerman (collaboration with Villén lab)
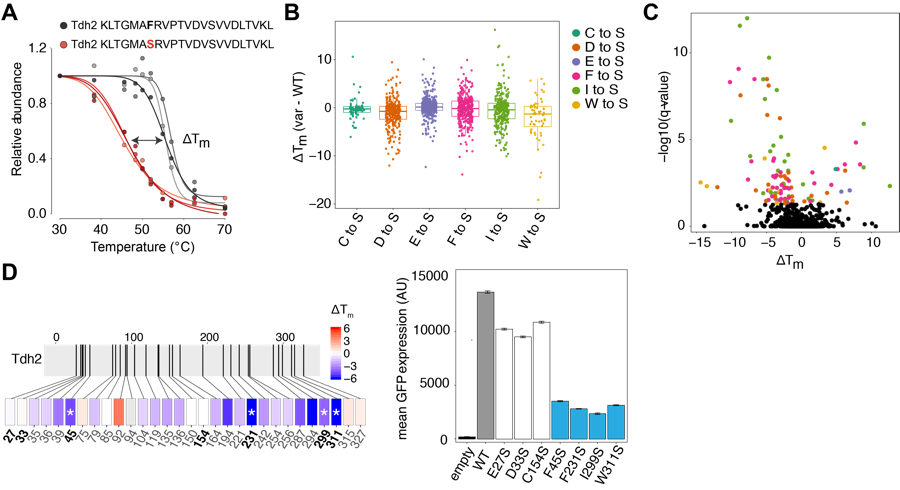
To measure the thermal stability of thousands of variant proteins in parallel, we used mistranslation coupled to the thermal proteome profiling method (TPP) (Savitski et al. 2014). In a TPP experiment, cells are incubated at increasing temperatures spanning the melting point (Tm) of most proteins and then lysed, and the soluble fraction of the proteome is collected. Each temperature point is labeled with an isobaric tandem mass tag (TMT), combined, and analyzed by MS. The TMTs allow a quantitative comparison of each temperature condition in the same MS run. As proteins denature and misfold at higher temperatures, they become depleted in the soluble fraction. Measuring the relative intensity of each peptide at each temperature allows the Tm to be calculated.
We used TPP on mistranslated proteomes in yeast to identify substitutions that alter protein thermal stability. We performed TPP on lysates from cells expressing one of six mistranslating tRNAs that substitute Ser at sites of 6 different amino acids and calculated the change in Tm between the wild type peptide and Ser-substituted peptides. In total, we measured Tm differences for ~1700 serine substitutions in ~400 proteins. We found that 14% of variants significantly altered thermal stability, most of which were destabilizing, and validated that variants that significantly reduce Tm reduce protein abundance.
We used TPP on mistranslated proteomes in yeast to identify substitutions that alter protein thermal stability. We performed TPP on lysates from cells expressing one of six mistranslating tRNAs that substitute Ser at sites of 6 different amino acids and calculated the change in Tm between the wild type peptide and Ser-substituted peptides. In total, we measured Tm differences for ~1700 serine substitutions in ~400 proteins. We found that 14% of variants significantly altered thermal stability, most of which were destabilizing, and validated that variants that significantly reduce Tm reduce protein abundance.
The yeast mating pathway as a model for complex trait genetics
Stephanie Zimmerman, Michael Dorrity, and Lucia Shumaker (Collaboration with the Queitsch Lab, University of Washington Department of Genome Sciences)
Genome-wide association studies have identified many variants associated with complex traits, but these variants frequently explain only a small fraction of a trait’s heritability. Hypotheses to explain this “missing heritability” include the additive action of many individually small-effect variants or significant gene-by-gene and gene-by-environment interactions. We are using the yeast mating pathway as a model complex trait to experimentally measure the impact of additive and epistatic genetic variation on mating.
We performed individual deep mutational scanning experiments to profile the effects of thousands of mutations in three mating pathway genes encoding physically interacting proteins: STE5 (scaffold protein), STE11 (MAPKKK), and STE7 (MAPKK). For each gene, we transformed the corresponding yeast deletion strain with a library of ~50,000 variants, carried out a selection for mating, and sequenced the population of variants before and after selection by high-throughput sequencing. Variants that lead to impaired mating function are depleted in the population after the selection, while variants that lead to wild-type mating are enriched. From these experiments, we identified mutations in all three genes across the full spectrum of effect sizes, from complete loss-of-function at key residues to many neutral to slightly deleterious mutations.
We performed individual deep mutational scanning experiments to profile the effects of thousands of mutations in three mating pathway genes encoding physically interacting proteins: STE5 (scaffold protein), STE11 (MAPKKK), and STE7 (MAPKK). For each gene, we transformed the corresponding yeast deletion strain with a library of ~50,000 variants, carried out a selection for mating, and sequenced the population of variants before and after selection by high-throughput sequencing. Variants that lead to impaired mating function are depleted in the population after the selection, while variants that lead to wild-type mating are enriched. From these experiments, we identified mutations in all three genes across the full spectrum of effect sizes, from complete loss-of-function at key residues to many neutral to slightly deleterious mutations.
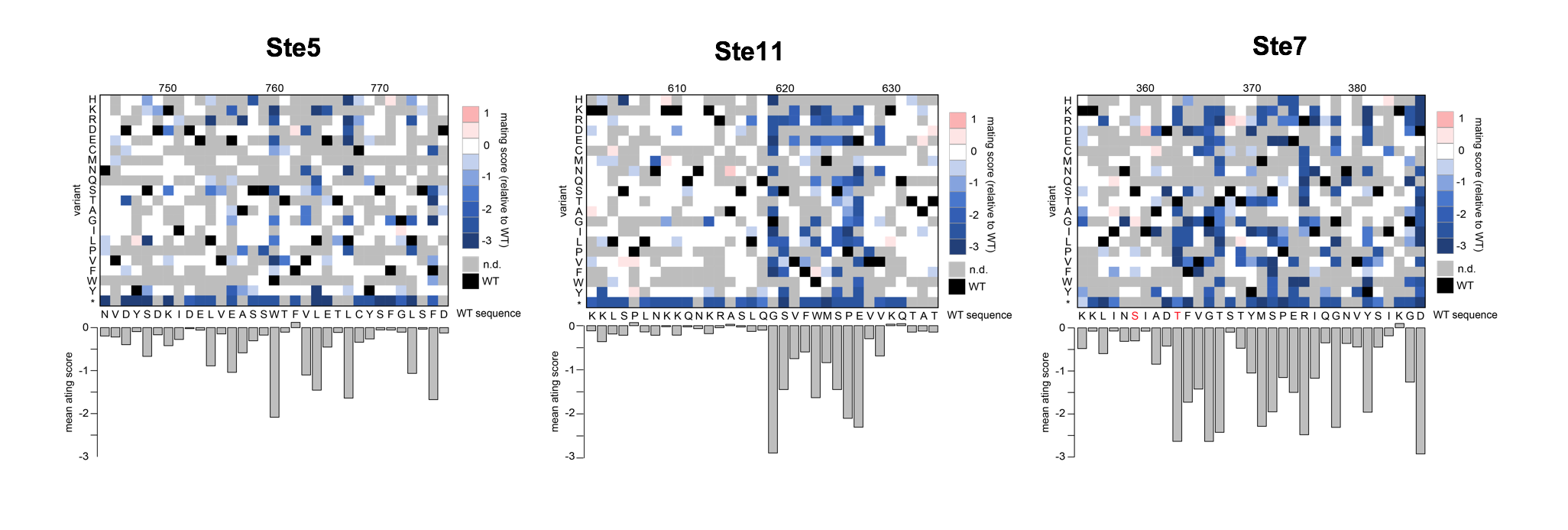
From the mutational scanning data, we selected 100 mutations in each gene with small to intermediate effects on mating and constructed libraries with combinations of these mutations found in two genes (10,000 combinations per gene pair). We transformed these libraries into yeast deleted for both genes to perform selections to measure the effect on mating of each double mutant. From these experiments, we will determine how well different models of genetic interaction predict the behavior of double mutants and comprehensively assess the relative impact of additive and epistatic interactions.
Engineering glyphosate-resistant cassava
Josh Cuperus and Ben Brandsen
Glyphosate, the active ingredient in Roundup, is a widely used, broad spectrum herbicide. It inhibits the enzyme 5-enolpyruvylshikimate-3-phosphate synthase (EPSPs), which is part of the shikimate pathway and needed to produce tyrosine, tryptophan, and phenylalanine. Thus, glyphosate inhibits the growth of sensitive plants.
We intend to develop a glyphosate-resistant variety of cassava, an important staple crop in sub-Saharan Africa. Cassava cultivation is often constrained by weed proliferation, as more than 50% of cassava production costs are allocated to weeding. Multiple single and double amino acid substitutions in EPSPs are known to confer glyphosate resistance, but all result in a slower-growing plant. Our goal is to identify one or more mutations in the cassava EPSPs that confer resistance to glyphosate, but which retain activity similar to that of the wild-type enzyme.
We generated a library of EPSPs variants using error-prone PCR, and introduced these into an E. coli strain lacking its native EPSPs gene. In this background, complementation by the cassava EPSPs is required for growth in minimal media. We then performed a growth-based selection in various concentrations of glyphosate to identify mutations that improved glyphosate resistance (Figure 1). Using high-throughput DNA sequencing, we determined the fitness conferred by each EPSPs variant based on its change in frequency before and after glyphosate selection.
We measured the growth of the best-performing variants in our E. coli host, and identified one mutation that improves the growth of both the WT EPSPs as well as of the T181L P185A double mutant, but it fails to improve resistance in another glyphosate-resistant variant, EPSPs T181I P185M. We have preliminary data that suggest that this mutation improves glyphosate resistance in Cassava plants.
We have also identified a second mutation that improves resistance of both WT and the T181L P185A double mutant. It appears to act additively with the first mutation that confers glyphosate resistance, improving resistance even further. As a rapid method to assess glyphosate resistance in a plant host, we are now testing combinations of these mutations in Camelina sativa. We hope to observe growth of C. sativa when complemented with our engineered variants of Cassava EPSPs compared to wild type Cassava EPSPs.
We intend to develop a glyphosate-resistant variety of cassava, an important staple crop in sub-Saharan Africa. Cassava cultivation is often constrained by weed proliferation, as more than 50% of cassava production costs are allocated to weeding. Multiple single and double amino acid substitutions in EPSPs are known to confer glyphosate resistance, but all result in a slower-growing plant. Our goal is to identify one or more mutations in the cassava EPSPs that confer resistance to glyphosate, but which retain activity similar to that of the wild-type enzyme.
We generated a library of EPSPs variants using error-prone PCR, and introduced these into an E. coli strain lacking its native EPSPs gene. In this background, complementation by the cassava EPSPs is required for growth in minimal media. We then performed a growth-based selection in various concentrations of glyphosate to identify mutations that improved glyphosate resistance (Figure 1). Using high-throughput DNA sequencing, we determined the fitness conferred by each EPSPs variant based on its change in frequency before and after glyphosate selection.
We measured the growth of the best-performing variants in our E. coli host, and identified one mutation that improves the growth of both the WT EPSPs as well as of the T181L P185A double mutant, but it fails to improve resistance in another glyphosate-resistant variant, EPSPs T181I P185M. We have preliminary data that suggest that this mutation improves glyphosate resistance in Cassava plants.
We have also identified a second mutation that improves resistance of both WT and the T181L P185A double mutant. It appears to act additively with the first mutation that confers glyphosate resistance, improving resistance even further. As a rapid method to assess glyphosate resistance in a plant host, we are now testing combinations of these mutations in Camelina sativa. We hope to observe growth of C. sativa when complemented with our engineered variants of Cassava EPSPs compared to wild type Cassava EPSPs.
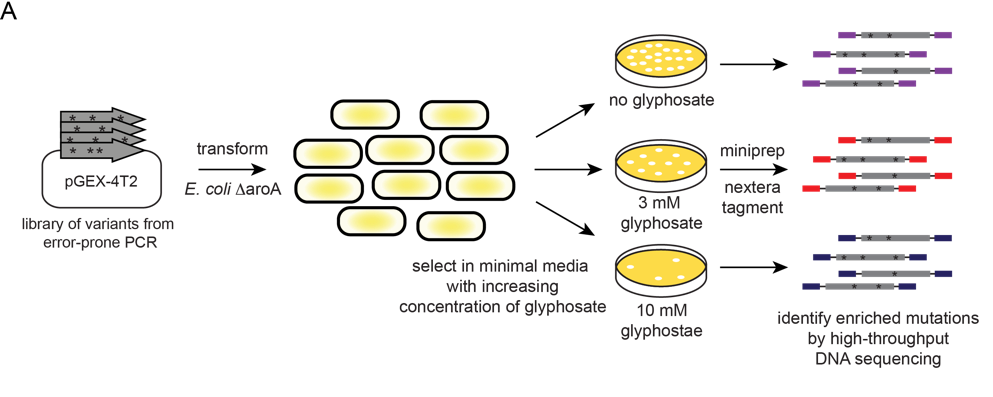
Figure 1. Experimental design to identify EPSPs variants with increased activity and glyphosate resistance. An error-prone PCR library was introduced into E. coli lacking EPSPs (aroA). These were plated on minimal media plates with various concentrations of glyphosate. After colonies were observed, the plates were scraped, EPSPs plasmid collected, and EPSPs gene prepared for deep-sequencing analysis with Nextera tagmentation.
Mutational scanning of an antibacterial peptide
Ethan Hills and Ben Brandsen
Antibiotic resistance is a growing threat to public health. This resistance, coupled with a dearth of new antibiotics, makes development of new antibiotics of critical importance. Many antibiotics derive from microbial pathways that synthesize complex natural products, and engineering these pathways to produce new antibiotics is an exciting prospect. We are using such a strategy to produce variants of the antibacterial lasso peptides Microcin J25, Klebsidin, and Acinetodin in E. coli. Named for their distinct lariat knot structure, each peptide is produced by three biosynthetic enzymes that modify and export a ribosomally-synthesized precursor peptide. The mature lasso peptide halts bacterial growth by inhibiting the uptake of nucleoside triphosphates (NTPs) into RNA polymerase. We plan to use an assay that relies on cellular growth to measure the activity of many lasso peptide variants in parallel, and to identify lasso peptide variants that overcome a known resistance mutation.
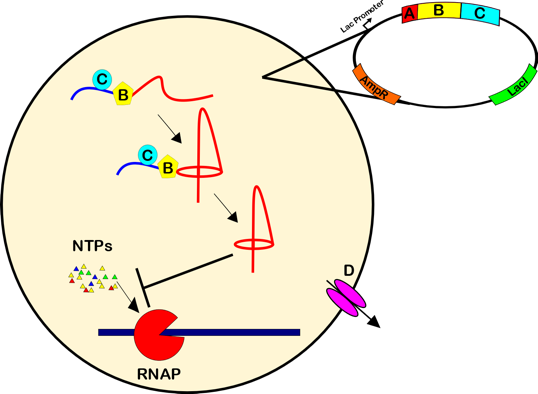
Figure 1. Lasso peptides Acinetodin, MccJ25, and Klebsidin are antibiotics that act by inhibiting transcription by obstructing the nucleoside triphosphate (NTP) uptake channel of RNA polymerase. Four proteins, denoted A, B, C, and D are involved in producing and exporting these peptides. The A protein encodes a leader peptide followed by the unprocessed core peptide. The B and C proteins post-translationally modify the A protein precursor to form the mature lasso structure. Finally, the D protein exports the lasso peptide outside of the cell. In our assays, the D gene was omitted to enable self-toxicity.
When the A, B, and C proteins for lasso peptide biosynthesis are expressed in E. coli, mature lasso peptides accumulate in the cell and inhibit growth. By expressing a library of lasso peptide variants, each within a single cell, and using deep sequencing to count the frequency of each variant before and after selection, we can obtain functional data for thousands of lasso peptides in one experiment. Using this assay, we hope to generate functional scores of each single amino acid mutation in Microcin J25, Klebsidin, and Acinetodin. These data will provide us an understanding of each lasso peptide’s tolerance to mutation and help us characterize the sequence of each peptide based on antibacterial activity. In addition, we hope to study how mutagenesis of the lasso peptides can combat antibiotic resistance. By performing the same selections in an E. coli strain resistant to the wild type lasso peptides, we hope to identify variants that overcome this resistance. Together, these studies will provide a framework for understanding the prospects of lasso peptide engineering and identifying novel peptide-based antibiotics.

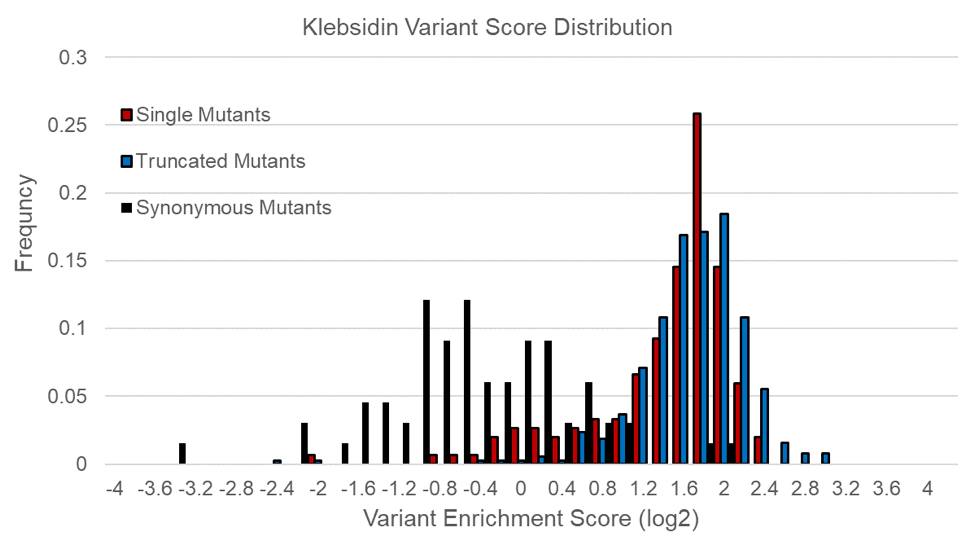
Figure 2. A. Wild Type lasso peptides Klebsidin and Microcin J25 successfully inhibit growth of the cell after induction with IPTG. B. Distribution of variant enrichment scores of the Klebsidin variant peptides normalized to the wild type score (zero). The truncated mutants show positive growth scores, indicating non-functional peptides. The single mutants show a slightly bimodal distribution with most non-functional, and a small population having neutral scores. The synonymous distribution is wider than anticipated, and indicates the selection requires optimization to move synonymous scores closer to zero. These data, while not yet ideal, show the selection is successfully separating Klebsidin variants based on antibacterial activity.
Dimensionality reduction by UMAP to visualize physical and genetic interactions
Mike Dorrity
Dimensionality reduction is often used to visualize complex expression profiling data. We used Uniform Manifold Approximation and Projection (UMAP) method on published transcript profiles of 1484 single gene deletions of Saccharomyces cerevisiae to identify layers of biological organization among these genes. Proximity in low-dimensional UMAP space identifies clusters of genes that correspond to protein complexes and pathways, and finds novel protein interactions even within well-characterized complexes. An example embedding of all 1484 genes is shown in Figure 1 below, where each gene is represented as a point, and clusters of points are grouped by color.
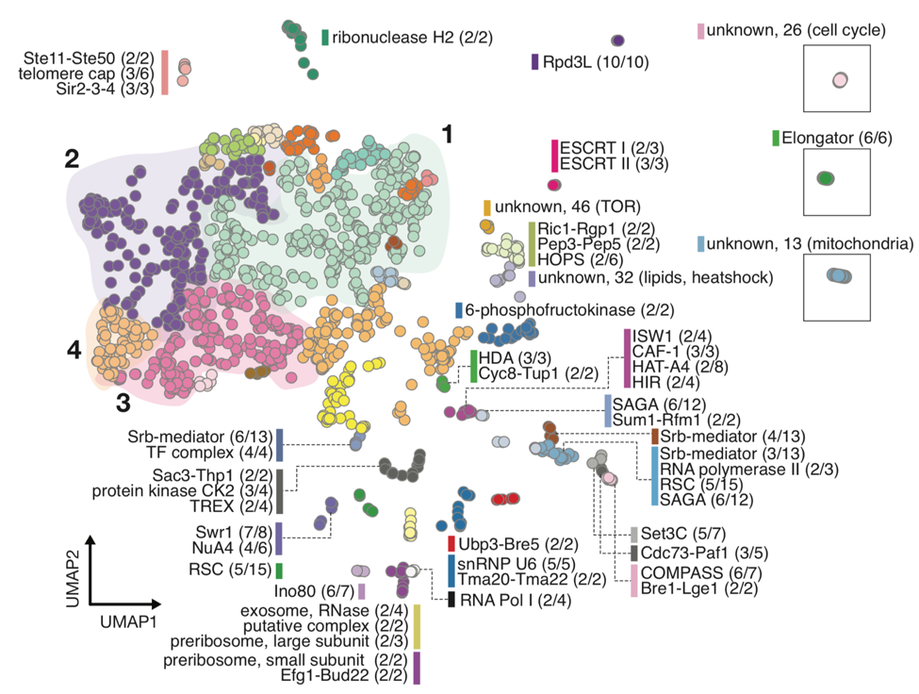
Figure 1.
This approach is more sensitive than previous methods and more effectively identifies true interactions defined by IP-mass spectrometry datasets. A ROC curve showing the increased efficacy of UMAP distance as a metric to predict true interactions is shown below in Figure 2.
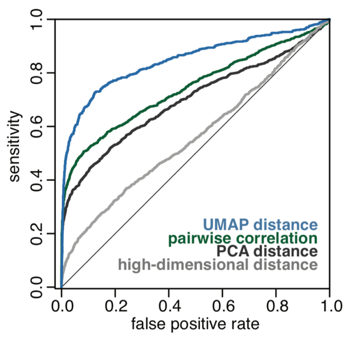
Figure 2.
An example of the method’s ability to pull out entire pathways is shown below in Figure 3. The tRNA wobble uridine pathway is captured entirely within the cluster containing the Elongator complex (boxed green cluster in Figure 1). Complex members within this cluster are annotated with orange boxes, while new members are annotated in blue. One pathway member, Nfs1, was not present in the single gene deletion dataset. The heatmap represents fine-scale distances between each pair of points within the cluster. Darker shades of red indicate points nearer in UMAP space. Heterodimeric interactions, such as Ncs6-Ncs2, are nearer to each other than other members of the pathway. Novel members of this pathway (blue text) are grouped with other members based on their similarity of UMAP distance, and these new interactions are indicated with gray lines in the pathway diagram. This approach should be broadly useful as additional transcriptome datasets become available for other organisms.
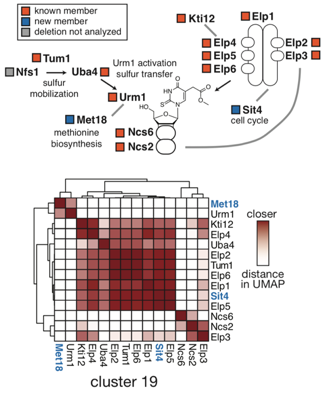
Figure 3.