Cis-elements that regulate protein expression in yeast
Molly Perchlik, Brooke Angell, Andrew Savinov, and Josh Cuperus
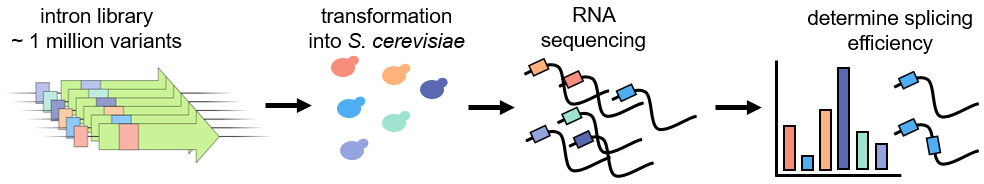
Non-coding cis-elements influence gene regulation. However, we still lack models that can effectively predict protein expression from an arbitrary non-coding sequence. One non-coding element, the 3′ untranslated region (3′ UTR), has an important role in transcription termination and mRNA processing and stability. To better understand the regulatory effects of the 3′ UTR and of other cis-elements in the yeast Saccharomyces cerevisiae, we constructed two libraries of 3′ UTRs consisting of 50 random nucleotides (N50) immediately downstream of the coding region of the HIS3 reporter gene. This approach analyzes millions of unique sequences, which provide far more data than libraries comprising the approximately 6,000 native yeast genes. Given that known RNA-protein binding and secondary structures motifs are relatively short, functional biological motifs should occur often in the N50 sequence.
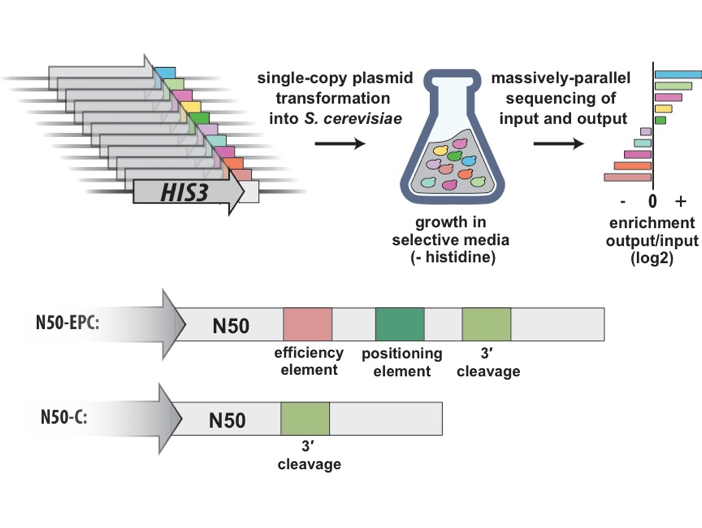
Studies in yeast have shown that a minimal terminator consisting of an efficiency element, positioning element, and cleavage site are sufficient to generate mature, polyadenylated transcripts. With this in mind, one library contains a 3′ cleavage site, efficiency element, and positioning element (N50-EPC) and the other contains only a 3′ cleavage site (N50-C). N50-EPC variants had a higher median growth rate than N50-C variants in media lacking histidine. In the N50-C context, for which the median activity of variants is low, elements that boost 3ʹ UTR activity were more apparent. Yeast transformants carrying a library member were subjected to a selection in which expression of the reporter gene correlates with growth in the absence of histidine. Deep sequencing of the DNA and mRNA from these libraries allowed us to survey the effects of sequence features and motifs on gene expression and mRNA cleavage position
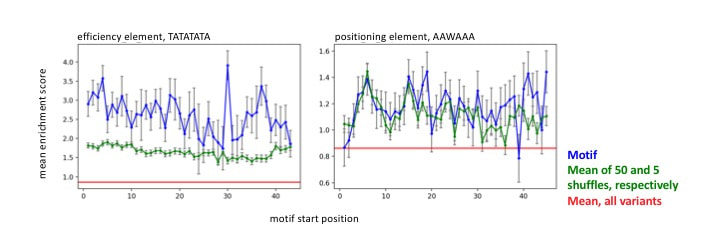
We observed motifs resembling efficiency and positioning elements in the N50-C library. The motif resembling the efficiency element, TATATATA, was enriched above 50 shuffled controls at almost every position in the N50. The motif resembling the positioning element, AAWAAA, was enriched above the average enrichment of all variants, but only enriched above shuffled controls at certain positions.
|
|
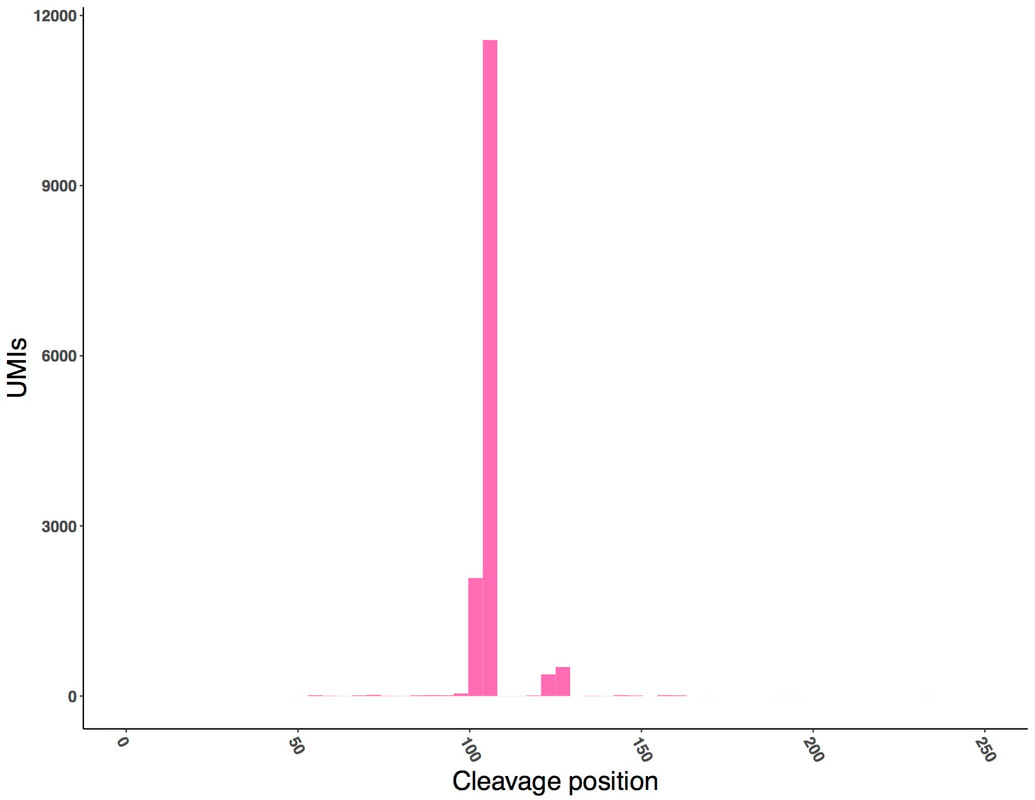
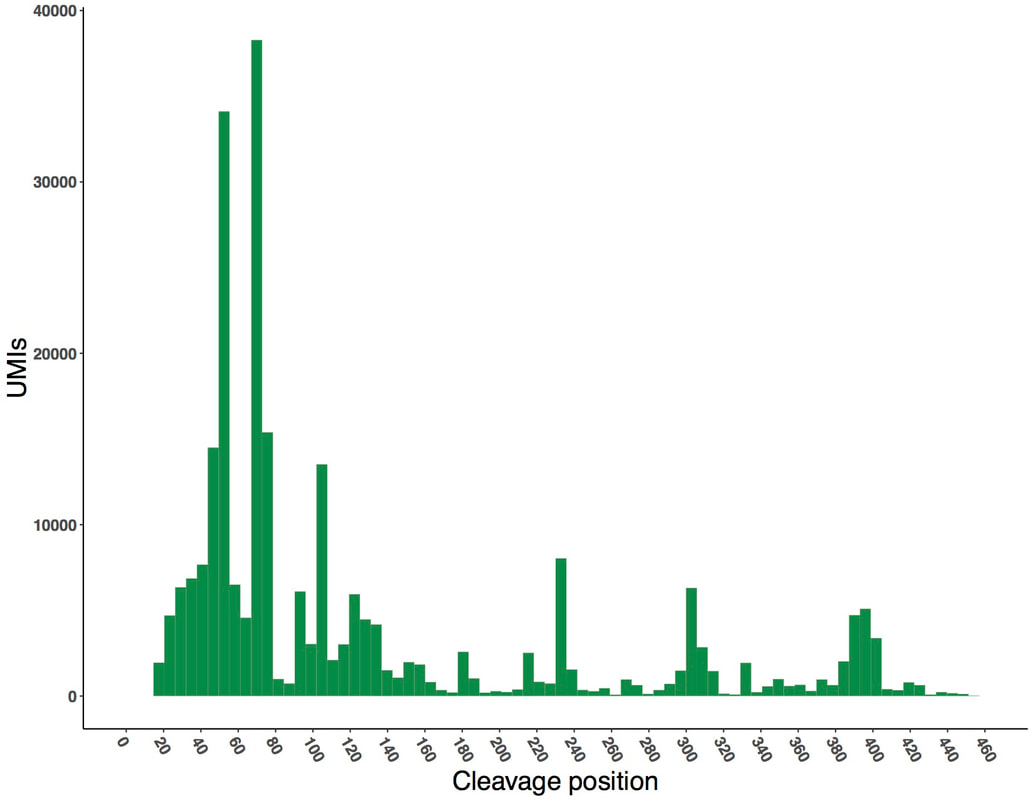
RNA-seq data was used to measure the cleavage sites of mRNA transcripts from both libraries. N50-EPC, containing an efficiency element and positioning element, had a nearly uniform 3ʹ cleavage site. Lacking these elements hard-coded into the library, N50-C variants showed greater variation in cleavage site position.
Intron sequences are another important type of non-coding cis-element. For many introns, splicing must occur to produce a functional mRNA, and thus the efficiency of splicing affects the total amount of protein produced. Introns have three key sequence elements – 5' and 3’ splice sites and a branch site – which are required for splicing. Further analyses of native intron sequences have identified additional enhancing and silencing motifs. However, much is still unknown about intron sequence motifs that influence splicing. To analyze a broader array of intron sequence variants yeast and to identify novel intron motifs that affect splicing, we constructed an intron library containing the three known splicing sequence elements as well as introducing ~ hundreds of thousands of variants of 50 random nucleotides and an identifying barcode in the 5’ UTR. We used RNA sequencing to determine splicing efficiency of this library and to identify cryptic splice events.
Single-cell genomics and development
Michael Dorrity, Joshua Cuperus, Ken Jean-Baptiste
|
|
Figure 1.
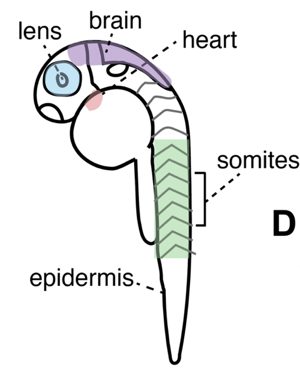
Single-cell transcriptomics allows for precise determination of individual cell types in a mixed cell population. We apply this technique to whole organisms during early development to identify transcriptional programs that promote development of specific cell lineages. In collaboration with Cole Trapnell’s lab, we use both microfluidic and combinatorial indexing approaches to generate single-cell molecular profiles. We focused specifically on enumerating cell lineages present in early development of the model plant Arabidopsis thaliana and the vertebrate Danio rerio (Figure 1). With this information, we hope to use both organisms to address the following questions: (1) What groups of genes promote specification of each cell lineage? (2) Can we identify rare cell types that were inaccessible to previous methods? (3) Can we pinpoint crucial gene regulatory events during development by combining single-cell expression (RNA-seq) with open chromatin (ATAC-seq) profiling techniques?
Single-cell transcriptomics allows for precise determination of individual cell types in a mixed cell population. We apply this technique to whole organisms during early development to identify transcriptional programs that promote development of specific cell lineages. In collaboration with Cole Trapnell’s lab, we use both microfluidic and combinatorial indexing approaches to generate single-cell molecular profiles. We focused specifically on enumerating cell lineages present in early development of the model plant Arabidopsis thaliana and the vertebrate Danio rerio (Figure 1). With this information, we hope to use both organisms to address the following questions: (1) What groups of genes promote specification of each cell lineage? (2) Can we identify rare cell types that were inaccessible to previous methods? (3) Can we pinpoint crucial gene regulatory events during development by combining single-cell expression (RNA-seq) with open chromatin (ATAC-seq) profiling techniques?
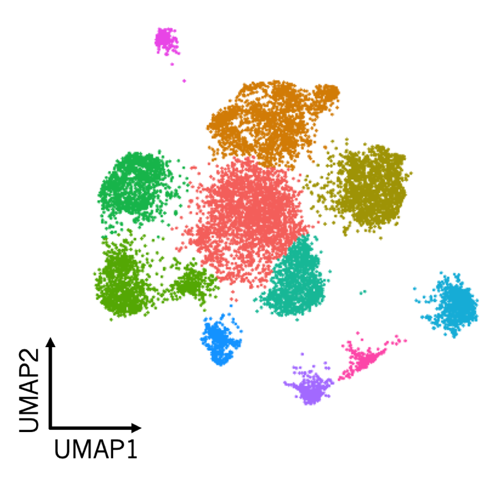
Figure 2.
We are in the initial stages of profiling 24-hour embryos of Danio rerio with scRNA-seq to characterize rare cell lineages present at this early developmental stage and to identify genes whose expression promotes specification of these lineages. In Arabidopsis thaliana, we have focused specifically on the development of roots, a tissue with crucial roles in nutrient uptake, water management, and response to stress. Using single-cell RNA-seq, we identified individual cell types that play important roles in these processes, and many new genes whose expression contribute to cell-type specific functions (Jean-Baptiste et al, 2019). We have further studied this tissue using single-cell ATAC-seq to identify regulatory regions underlying cell-type specific expression patterns. The above low-dimensional UMAP embedding of scATAC data (Figure 2) shows individual cells colored by shared cell type from the developing root of the model plant Arabidopsis thaliana. While we identify similar numbers of individual cell types using scATAC-seq, these methods find distinct molecular features: some cell type-specific open chromatin changes show no corresponding change in expression. Furthermore, some cell types show distinct subpopulations that are not present in our earlier scRNA-seq study
We are in the initial stages of profiling 24-hour embryos of Danio rerio with scRNA-seq to characterize rare cell lineages present at this early developmental stage and to identify genes whose expression promotes specification of these lineages. In Arabidopsis thaliana, we have focused specifically on the development of roots, a tissue with crucial roles in nutrient uptake, water management, and response to stress. Using single-cell RNA-seq, we identified individual cell types that play important roles in these processes, and many new genes whose expression contribute to cell-type specific functions (Jean-Baptiste et al, 2019). We have further studied this tissue using single-cell ATAC-seq to identify regulatory regions underlying cell-type specific expression patterns. The above low-dimensional UMAP embedding of scATAC data (Figure 2) shows individual cells colored by shared cell type from the developing root of the model plant Arabidopsis thaliana. While we identify similar numbers of individual cell types using scATAC-seq, these methods find distinct molecular features: some cell type-specific open chromatin changes show no corresponding change in expression. Furthermore, some cell types show distinct subpopulations that are not present in our earlier scRNA-seq study
Expanding binding specificity for RNA recognition by a Puf domain
Wei Zhou
The ability to design a protein that can bind specifically to any RNA and regulate its fate would enable numerous research and therapeutic applications. However, decoding the RNA-binding specificity for most types of RNA-binding domains is challenging, as these domains associate with RNA via complex networks of interactions. The modular architecture of the Puf domain, with eight repeats that each contact one base, make this domain an ideal candidate to generate new RNA-binding specificities (Figure 1A, 1B). For each repeat of the Pumilio-1 Puf domain, we generated a library of variants that contains the 8,000 possible combinations of amino acid substitutions at residues 12, 13 and 16 that are critical for RNA contact. We then carried out yeast three-hybrid selections with each library against the RNA recognition sequence for the Pumilio-1 domain, with any possible base present at the cognate position recognized by the repeat that was randomized (Figure 1C). We used next generation sequencing technology to score the binding activity of each variant for its ability to bind to each possible RNA base at the cognate position. We assigned a specificity and affinity score for each variant by comparing its binding across the four RNA bases, identifying many variants with highly specific interactions (Figure 2). This approach allows us to propose a complete code for RNA recognition by this domain.
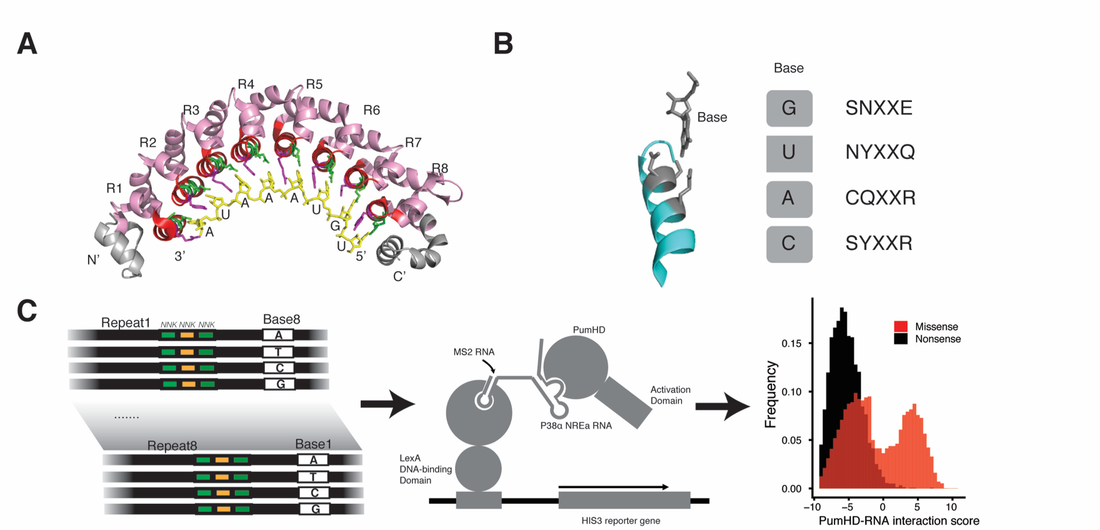
Figure 1: The yeast three-hybrid method is combined with next generation sequencing to score the binding activities of each mutated PumHD repeat to each of the 4 possible RNA bases. (A) Illustration of PumHD domain structure. (B) Base-specific binding of PumHD. (C) Schematic flow of library design, selection and sequencing.
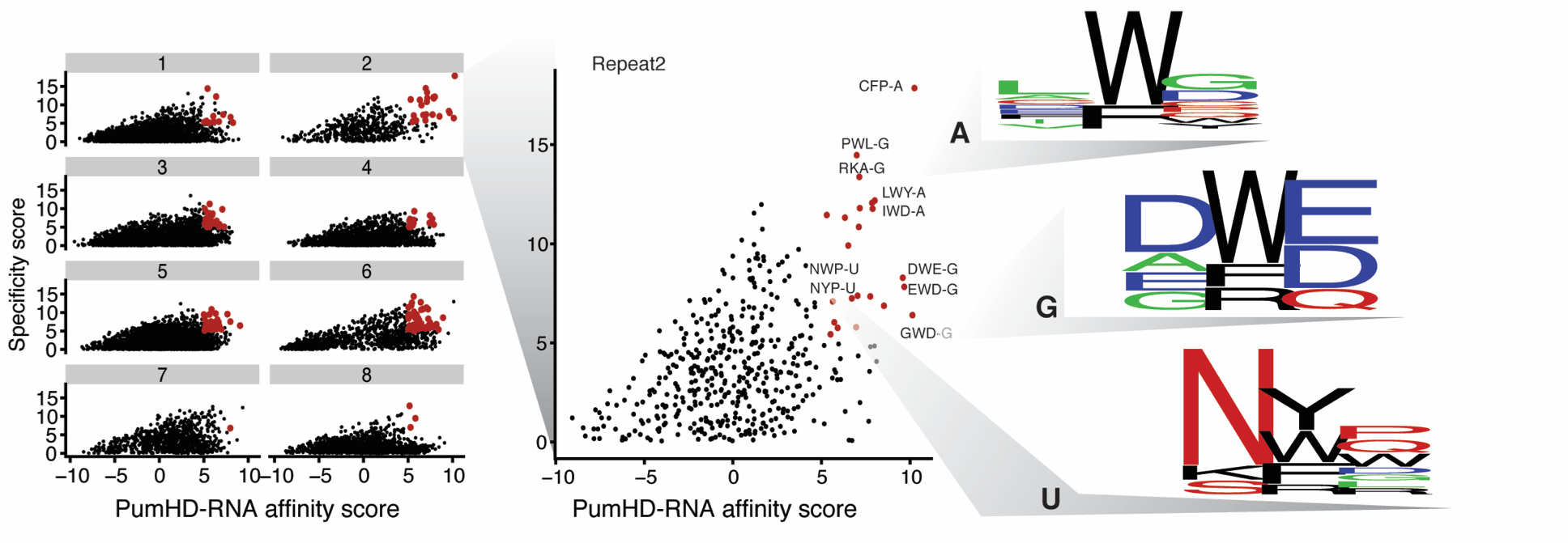
Figure 2: The specificity and affinity of each PumHD variant for each RNA base.