DNA Technology
Empirical Estimation of Robustness from Deep Mutational Scans
Bryan Andrews (Collaboration with Peter Conlin and Ben Kerr
Are genes more robust to errors than expected by chance?
Because of the way the genetic code is structured, different codons for the same amino acid have different ‘mutational neighborhoods.’ That is, if a single-base substitution occurs at an arginine codon, for instance, the set of possible new amino acids is very different if the original codon was an ‘cgg’ versus an ‘agg’, and the same is true for half of the amino acids: A, G, I, K, L, P, R, S, T & V.
Because of the way the genetic code is structured, different codons for the same amino acid have different ‘mutational neighborhoods.’ That is, if a single-base substitution occurs at an arginine codon, for instance, the set of possible new amino acids is very different if the original codon was an ‘cgg’ versus an ‘agg’, and the same is true for half of the amino acids: A, G, I, K, L, P, R, S, T & V.
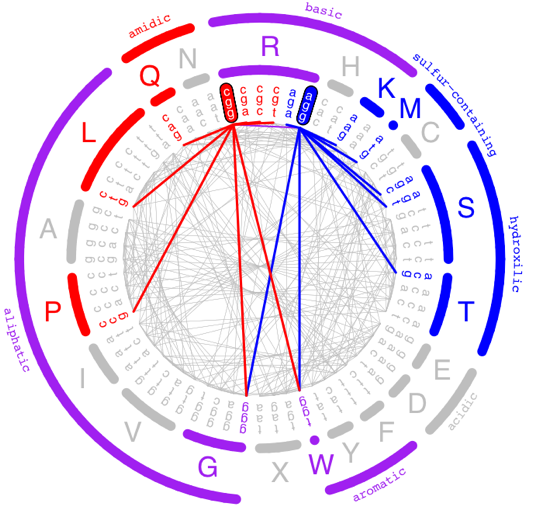
As a result, the potential effects of a single-base substitution on a protein depends on what codons are used to encode that protein. In this project, in collaboration with Peter Conlin and Ben Kerr, we computationally reanalyzed published datasets in which proteins were subjected to Deep Mutational Scans, in order to test whether naturally occurring gene sequences are more robust to errors than would be expected by chance, a property we are calling ‘genetic robustness.’
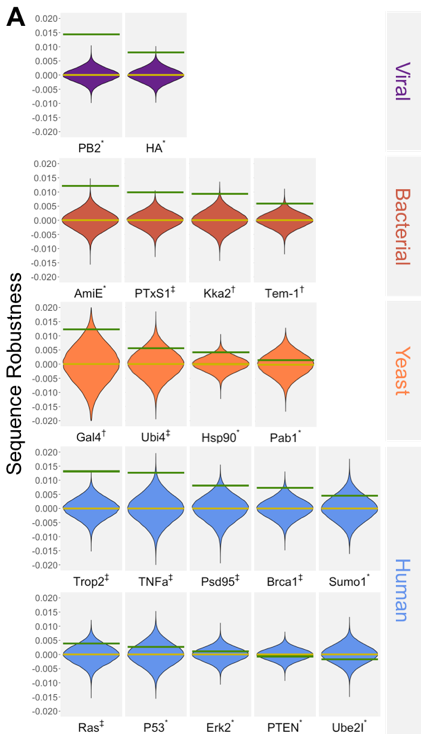
In the above figure, we compare the robustness of the wild type sequences (green lines) to a distribution of 100,000 sequences encoding the same protein with randomly selected codons. With a few exceptions, we see broad evidence that actual genes are more robust than synonymous random encodings. Amino acids that are buried, that are empirically sensitive to mutation, and that are evolutionarily conserved all appear to be more robust than sites that tolerate mutations. Furthermore, gene-wide codon frequencies are not sufficient to explain the degree of robustness observed; most of the signal comes from where specific codons are used rather than how frequently they are used. We posit the existence of an evolutionary mechanism to select for sequences that are robust to errors, but cannot distinguish with our data whether selection acts to remove sequences that give rise to inherited errors (i.e. rare low-fitness offspring), or non-inherited errors such as mistranslation events.
A processive CRISPR-guided base editing system
Ruth Groza
Efficient and precise in vivo mutagenesis of nucleic acids has been a longstanding objective of genetic engineering. Clustered regularly interspersed short palindromic repeat (CRISPR) guided base editing, a technology that couples Cas9 nucleases to nucleotide-modifying enzymes, offers a promising method of achieving that goal. However, current CRISPR-guided editors are limited to targeting regions within 350 nucleotides of protospacer adjacent motifs (PAMs), hampering their ability to diversify full-length genes (Halperin, S.O. et al., Nature 2018). We are seeking to develop a base editing system in S. cerevisiae that addresses this issue by combining the high processivity of a DNA helicase with the targeting precision of CRISPR, precluding the need for more expensive and labor-intensive gene tiling.
Our system hinges upon the recruitment of a chimeric protein to Cas9. In this chimera, a processive DNA modifying enzyme is fused to a base editing enzyme that is itself fused to the MS2 phage coat protein. Recruitment of the chimera to the target site is achieved via the interaction of the MS2 coat protein with MS2 RNA hairpins incorporated into the single guide RNA (sgRNA). In addition to recruiting the chimera, the sgRNA activates Cas9 and specifies its genomic target site. After initiating at the target site, the processive DNA modifying enzyme should move along the DNA in concert with the fused base editor as it installs mutations.
Our system hinges upon the recruitment of a chimeric protein to Cas9. In this chimera, a processive DNA modifying enzyme is fused to a base editing enzyme that is itself fused to the MS2 phage coat protein. Recruitment of the chimera to the target site is achieved via the interaction of the MS2 coat protein with MS2 RNA hairpins incorporated into the single guide RNA (sgRNA). In addition to recruiting the chimera, the sgRNA activates Cas9 and specifies its genomic target site. After initiating at the target site, the processive DNA modifying enzyme should move along the DNA in concert with the fused base editor as it installs mutations.
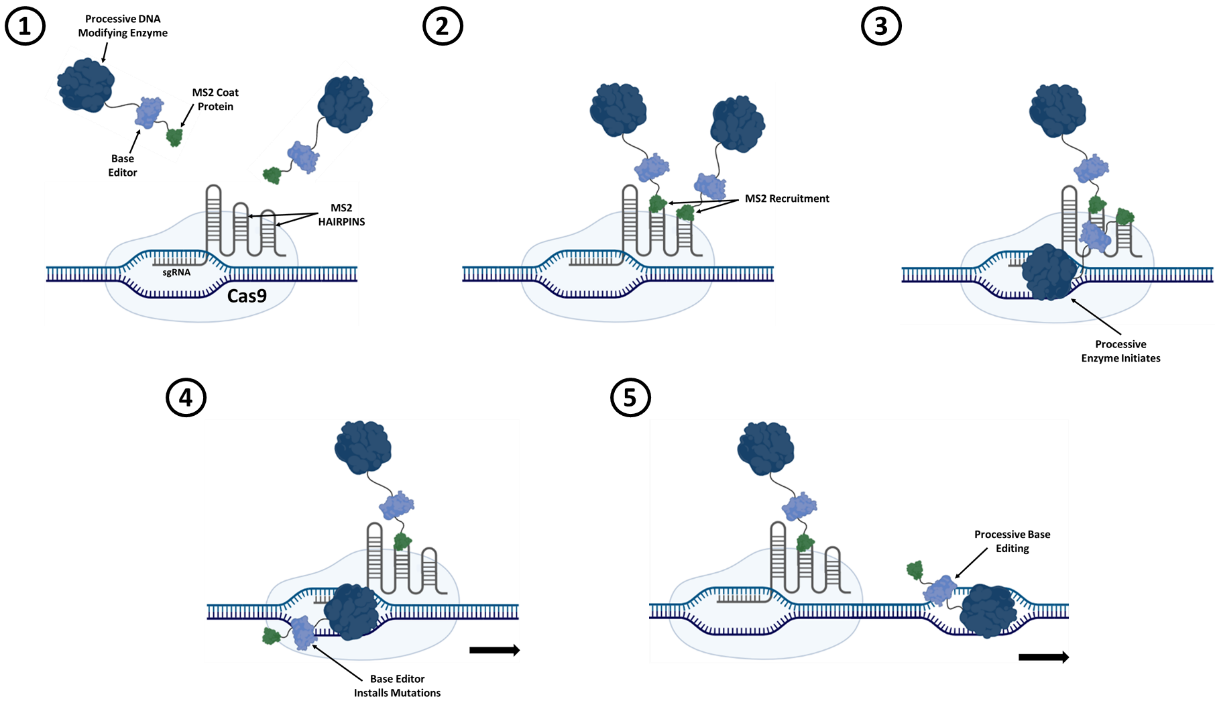
Figure 1. Schematic depicting a processive CRISPR-guided base editing system. A fusion of a processive DNA modifying enzyme, a base editor, and an MS2 coat protein is used to mutagenize DNA downstream of the Cas9 target site.
A tethering assay to analyze chromosome structure
Taylor Wang (former lab member)
Within the nucleus, chromosomes fold into complex, 3-dimensional structures that play a vital role in gene regulation. These structures include chromosome territories defined by physical organization, as well as both long- and short-range interactions between and within chromosomes. The biological importance of chromosomal organization has been established, but the causal relationship between cellular fitness and chromosomal interactions is still unclear. This causal relationship represents unknown territory that can be explored by methods that can artificially manipulate chromosomes to create new interactions and study their effects on fitness.
We use a novel assay that manipulates chromosomal interactions by taking advantage of Cas9 targeting flexibility from the clustered regularly interspersed short palindromic repeats (CRISPR) system for genome editing. Guide RNAs (gRNAs) provide the basis for flexibility by targeting Cas9 binding in the genome through sequence complementarity. I adapted this flexibility for my needs by using a nuclease deficient Cas9 (dCas9) that binds without making incisions. This dCas9 is expressed as a fusion to the sequence specific DNA-binding domain (DBD) of LexA. The LexA-dCas9 fusion creates tethers by bringing an anchor locus—containing the lexA operator sequence—into close proximity of the dCas9 binding site (Figure 1A). These tethers can be created in multiplex wherein each cell in a population receives a different gRNA library member and generates a unique tether. A fitness selection for tethering will result in changing gRNA frequencies which can be read out by Illumina sequencing (Figure 1B).
We use a novel assay that manipulates chromosomal interactions by taking advantage of Cas9 targeting flexibility from the clustered regularly interspersed short palindromic repeats (CRISPR) system for genome editing. Guide RNAs (gRNAs) provide the basis for flexibility by targeting Cas9 binding in the genome through sequence complementarity. I adapted this flexibility for my needs by using a nuclease deficient Cas9 (dCas9) that binds without making incisions. This dCas9 is expressed as a fusion to the sequence specific DNA-binding domain (DBD) of LexA. The LexA-dCas9 fusion creates tethers by bringing an anchor locus—containing the lexA operator sequence—into close proximity of the dCas9 binding site (Figure 1A). These tethers can be created in multiplex wherein each cell in a population receives a different gRNA library member and generates a unique tether. A fitness selection for tethering will result in changing gRNA frequencies which can be read out by Illumina sequencing (Figure 1B).
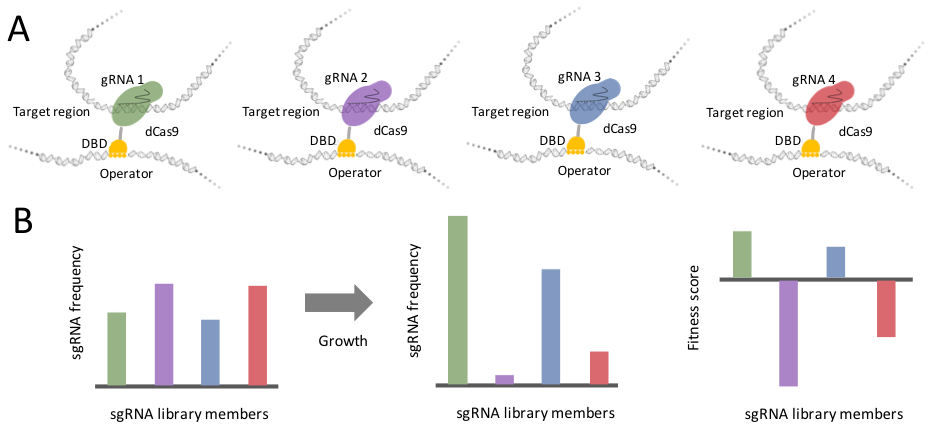
Figure 1. sgRNA library members result in unique tethers with varying effects on fitness.
(A) Schematic depicting four different tethers in cells with dCas9 binding sites indicated by colors. (B) Histograms describe expected results for changing frequencies of sgRNAs depending on their tethering effect. These changes in frequencies can be used to calculate fitness scores.
(A) Schematic depicting four different tethers in cells with dCas9 binding sites indicated by colors. (B) Histograms describe expected results for changing frequencies of sgRNAs depending on their tethering effect. These changes in frequencies can be used to calculate fitness scores.
In pilot experiments, we have been testing the efficacy of the tethering technology by tethering a plasmid-to- genome—instead of genome-to-genome—and using mCherry fluorescence as a reporter for the presence or absence of a tether. This plasmid-to-genome tethering experiment relied on three components: 1] exogenous constitutively expressed Gal4 DBD-VP16; 2] the LexA-dCas9 tethering protein; and 3] another exogenous plasmid that is bound by the dCas9 of the tethering protein and that also contains Gal4 binding sites (Figure 2). With this design we can test our hypothesis of trans-activator diffusion from one tethered locus to the other. If one of these loci is the reporter locus, this diffusion should allow for transcriptional activation of mCherry.
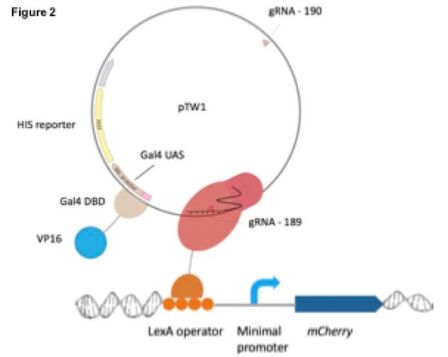
Moving forward, we also intend to adapt this system for genome-to-genome tethering at a single site that will act like pTW1 by binding the Gal4 DBD-VP16 activator, i.e. the Gal4 UAS on the pTW1 plasmid will instead be moved to a genomic locus. gRNA design will target the dCas9 to this genomic locus instead of the plasmid, and tethering of this locus to the reporter should result in activation if genome-to-genome tethering is possible. These experiments would open the door to the feasibility of multiplex library tethering experiments to investigate the effect of perturbing genomic organization on cellular fitness.
High throughput functional analysis of the fitness landscape of a yeast promoter
Matt Rich (former lab member) (with Celia Payen and Maitreya Dunham)
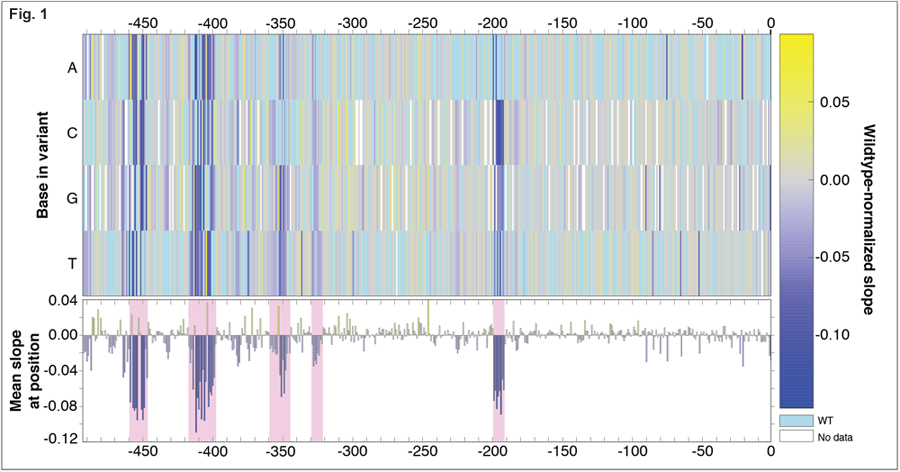
There are multiple ways to increase the expression of a gene, including mutations in cis-regulatory regions and gene amplification. When experimentally-evolved in limited sulfur, yeast reproducibly amplify the locus containing the gene for the high-affinity sulfur transporter, SUL1, increasing Sul1 protein expression. Neither coding nor cis-regulatory mutations have been found in evolved populations. To determine whether non-coding mutations are a viable evolutionary strategy to increase expression of SUL1, we created nearly 100,000 variants of the gene’s ~500 bp promoter and selected the resulting strains for fitness during sulfur limitation.
Most mutations to the SUL1 promoter do not have a significant effect on fitness. Fitness data for single mutants (Figure 1) map functional regions of the promoter (marked in pink), including the TATA box. Using our data, we can identify the transcription factors binding to sensitive regions as Cbf1 and Met32. The wildtype sequences in these regions are not the consensus site for each factor; mutations changing each sequence to the consensus binding site yield 5-10% increases in cellular fitness. We can also identify mutations that create new sites for Cbf1 and Met31 that also lead to 5-10% fitness increases.
Most mutations to the SUL1 promoter do not have a significant effect on fitness. Fitness data for single mutants (Figure 1) map functional regions of the promoter (marked in pink), including the TATA box. Using our data, we can identify the transcription factors binding to sensitive regions as Cbf1 and Met32. The wildtype sequences in these regions are not the consensus site for each factor; mutations changing each sequence to the consensus binding site yield 5-10% increases in cellular fitness. We can also identify mutations that create new sites for Cbf1 and Met31 that also lead to 5-10% fitness increases.
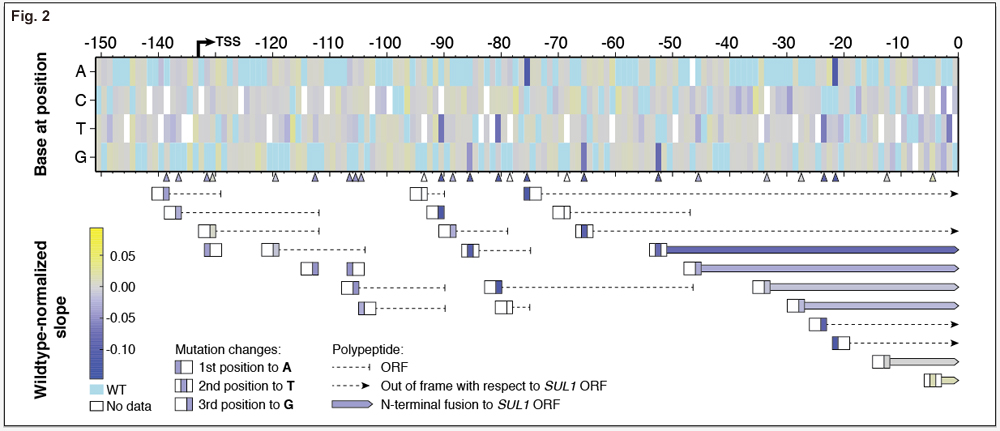
Selection in sulfur-limited chemostats is very sensitive and measures Sul1 protein levels in the cell. As such, we can also measure the effect of possible post-transcriptional regulation of SUL1, mainly through the creation of upstream open reading frames (uORFs) in the 5’ untranslated region (Figure 2). New uORFs have little effect if they are not within the 100 bases upstream of the SUL1 start codon. Within 100 bases, most uORFs decrease fitness. The longest N-terminal fusion initiated at a uORF is 14 amino acids and leads to ~10% decrease in cellular fitness. As these fusions decrease in length, effects on cellular fitness become gradually more neutral.
DNA shuffling methods for identify functional protein residues
Matt Rich (former lab member)
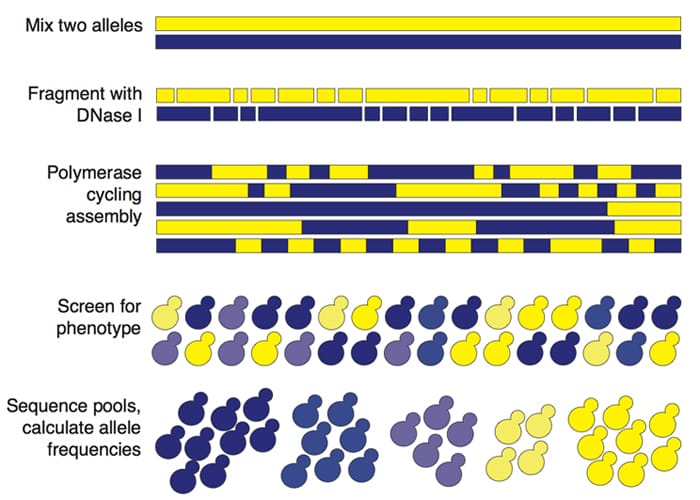
DNA shuffling is a method by which similar sequences are fragmented and reassembled to create chimeric versions of the two sequences. These chimeras can be screened or selected for function and sequenced, allowing the identification of the residues responsible for the phenotype of interest by analyzing allele frequencies. We are applying these methods in yeast to study protein co-evolution with regard to the competition between virus and host defense, and mapping quantitative trait loci at the level of functional nucleotides.
Human cells employ various mechanisms to combat viral infection. For instance, in response to double-stranded RNA, protein kinase R (PKR) stops translation by phosphorylating the translation initiation factor EIF2a. To avoid this cellular response, poxviruses express proteins that mimic EIF2a, like the vaccinia protein K3L, and PKR phosphorylates these instead of EIF2a. Elde et al. (Nature 2009) showed that these proteins are under fast positive selection, and that there is a differential response across the primate lineage for response to EIF2a mimics. Elde et al. defined the residues in PKR that are responsible for the divergence between human, gibbon, and orangutan PKR using yeast expression to measure the effect of K3L on PKR. We are coupling DNA shuffling to further map the competition between K3L and PKR, first by shuffling the human and gibbon PKRs and comparing our results after screening for K3L evasion.
We are also applying DNA shuffling to finely map quantitative trait loci. After coarsely mapping QTLs, a number of further genetic analyses -- like reciprocal hemizygosity mapping, allele swapping, and site-directed mutagenesis – are usually used to finely map the locus to identify the specific variants causing the phenotype. Expressing a library of chimeric sequences in a knockout strain should accomplish most of these analyses. We are currently applying this methodology to a mapped QTL for ammonium toxicity.
Human cells employ various mechanisms to combat viral infection. For instance, in response to double-stranded RNA, protein kinase R (PKR) stops translation by phosphorylating the translation initiation factor EIF2a. To avoid this cellular response, poxviruses express proteins that mimic EIF2a, like the vaccinia protein K3L, and PKR phosphorylates these instead of EIF2a. Elde et al. (Nature 2009) showed that these proteins are under fast positive selection, and that there is a differential response across the primate lineage for response to EIF2a mimics. Elde et al. defined the residues in PKR that are responsible for the divergence between human, gibbon, and orangutan PKR using yeast expression to measure the effect of K3L on PKR. We are coupling DNA shuffling to further map the competition between K3L and PKR, first by shuffling the human and gibbon PKRs and comparing our results after screening for K3L evasion.
We are also applying DNA shuffling to finely map quantitative trait loci. After coarsely mapping QTLs, a number of further genetic analyses -- like reciprocal hemizygosity mapping, allele swapping, and site-directed mutagenesis – are usually used to finely map the locus to identify the specific variants causing the phenotype. Expressing a library of chimeric sequences in a knockout strain should accomplish most of these analyses. We are currently applying this methodology to a mapped QTL for ammonium toxicity.
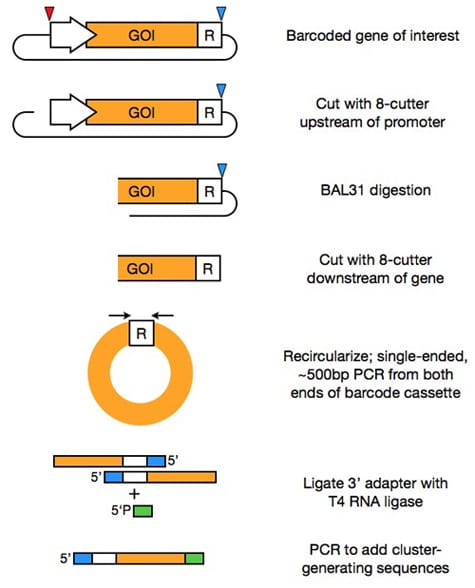
Length-agnostic, barcode-directed assembly of gene haplotypes
Matt Rich (former lab member)
Barcoding and in silico assembly of mutagenized sequences has expanded the scale at which DNA sequences can be functionally assayed, as sequence spanning multiple short, high-throughput sequencing reads can be assembled into a single allele using unique barcodes (Patwardhan, Hiatt, et al. Nat Biotechnology (2012)). Although this method enabled the resolution of haplotypes of sequences of up to approximately one kilobase (the cluster-generating limit of an Illumina sequencer), current techniques require further molecular biological manipulations, such as the combinatorial removal of internal regions, to resolve the haplotypes of longer sequences. We are developing a method for barcoded assembly of sequences that circumvents this 1 kb limit. We linearize the plasmid containing our barcoded gene of interest upstream of the gene's promoter, then use an endonuclease to digest the fragment from each end. After removing the remaining plasmid backbone, we are left with many barcoded fragments of different lengths. Recircularization brings the endonuclease-digested end of the gene proximal to the barcode. Single-ended, short PCR followed by single-stranded ligation creates sequences that can be clustered and sequenced on an Illumina high-throughput sequencer. These reads can then be merged by their barcode sequences. We will apply this technique first to the selections for the function of large, mutagenized yeast transcription factors, like those encoded by the ADR1 and MSN2 genes, but the technique should be generalizable to many applications that require the resolved haplotypes of large genes.
Transcriptional engineering of ethanol-tolerant yeast strains
Matt Rich (former lab member)
Alcohols cause pleiotropic cellular stress by disrupting the cell membrane and non-specifically destabilizing proteins. In yeast over 1000 genes have been implicated in increasing alcohol tolerance. Given such complexity, methods like transcriptional engineering that modulate cellular processes genome-wide are ideal tools to analyze this trait. In 2006, Alper and colleagues showed that variants of the yeast TATA-binding protein (Spt15) could improve viability at 6% ethanol. Spt15 regulates the expression of nearly all genes, so while its variants modulate many genes that are necessary for alcohol tolerance, they likely have off-target and possibly deleterious effects. To examine the possibility that variants of less ubiquitous transcription factors can also be used to increase ethanol tolerance, we created libraries consisting of over one million variants for three alcohol-responsive yeast transcription factors, Asr1, Msn2 and Msn4 and selected yeast containing these factors at 7.5% ethanol.
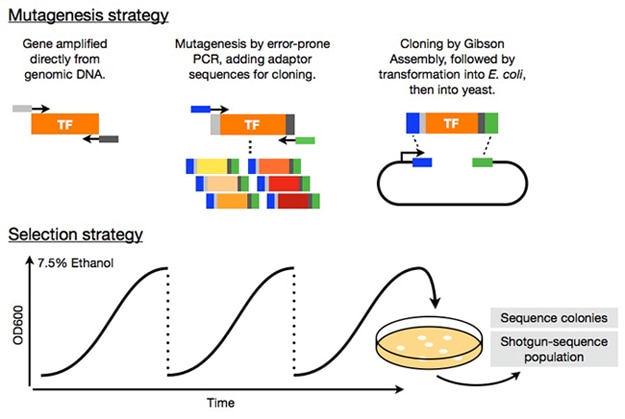
Many non-synonymous and frameshift mutations in the ASR1 and MSN genes enriched over the course of selection. We are continuing selections and confirming the tolerance of highly-enriched mutations. After this confirmation, we plan to use RNA-sequencing to analyze the transcriptional changes underlying the tolerance phenotype, in an effort to elucidate the molecular basis of yeast ethanol tolerance. We also believe that, if successful, this approach could be used to investigate the molecular basis of other complex traits.
Functional screening of soil metagenomic libraries
Kelly McGarvey (former lab member)
Most of the genomes of environmental microorganisms are inaccessible because they cannot be cultured in the lab by standard techniques. However, we can access the genomes of these unculturable organisms by extracting DNA directly from environmental samples and creating metagenomic DNA libraries. Using this approach, DNA from thousands of environmental microbes can be functionally screened for a variety of abilities. We are pursuing 2 different strategies to create and functionally screen environmental DNA libraries.
Using standard methods to functionally screen DNA libraries in an E. coli host, we are investigating the antibiotic resistance mechanisms coded in uncultured soil microorganisms. In particular, we have identified sequences from an environmental DNA library that allow growth of an E. coli host in the presence of 6 different antibiotics that function by targeting varied cellular pathways. We have found new sequences coding for many families of antibiotic resistance proteins including the antibiotic modifying enzymes rifampin ADP-ribosylases and aminoglycoside acetyltransferases, transporter proteins that are able to pump antibiotics out of the cell, as well as proteins like dihydrofolate reductases that are able to evade antibiotics when exogenously expressed in E. coli. We hope to use these new sequences to learn more about the evolution and functions of these protein families.
We are additionally interested in developing new ways to screen environmental DNA libraries in order to overcome the limitations associated with functional screening in a laboratory host. Standard screening requires that a heterologously expressed protein is functional in the host bacteria, and also requires the availability of an assay to test the function of interest on a large scale. Cloning an environmental DNA library into a phage backbone and screening the phage library via affinity selection would allow more permissive and efficient screening since protein domains that bind to a substrate of interest could be recovered without relying on the function of the encoded protein in a foreign host. This screening strategy will be widely applicable to a variety of binding and catalytic functions. We hope to use affinity selection of a metagenomic phage display library to search for antibiotic resistance proteins and inhibitors of these resistance proteins.
Using standard methods to functionally screen DNA libraries in an E. coli host, we are investigating the antibiotic resistance mechanisms coded in uncultured soil microorganisms. In particular, we have identified sequences from an environmental DNA library that allow growth of an E. coli host in the presence of 6 different antibiotics that function by targeting varied cellular pathways. We have found new sequences coding for many families of antibiotic resistance proteins including the antibiotic modifying enzymes rifampin ADP-ribosylases and aminoglycoside acetyltransferases, transporter proteins that are able to pump antibiotics out of the cell, as well as proteins like dihydrofolate reductases that are able to evade antibiotics when exogenously expressed in E. coli. We hope to use these new sequences to learn more about the evolution and functions of these protein families.
We are additionally interested in developing new ways to screen environmental DNA libraries in order to overcome the limitations associated with functional screening in a laboratory host. Standard screening requires that a heterologously expressed protein is functional in the host bacteria, and also requires the availability of an assay to test the function of interest on a large scale. Cloning an environmental DNA library into a phage backbone and screening the phage library via affinity selection would allow more permissive and efficient screening since protein domains that bind to a substrate of interest could be recovered without relying on the function of the encoded protein in a foreign host. This screening strategy will be widely applicable to a variety of binding and catalytic functions. We hope to use affinity selection of a metagenomic phage display library to search for antibiotic resistance proteins and inhibitors of these resistance proteins.
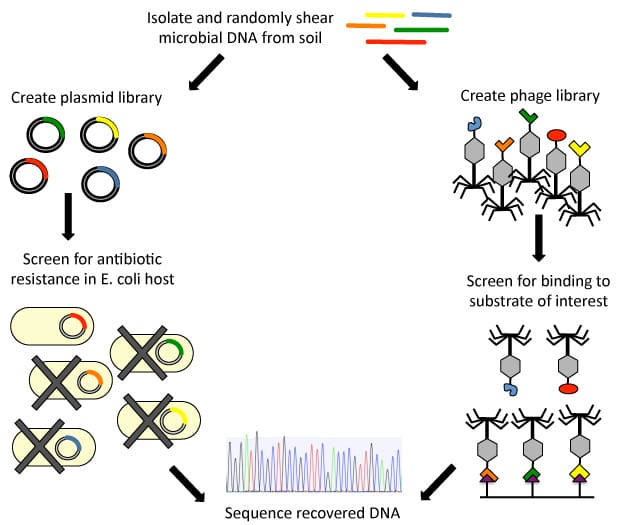
Figure 1. We are using two strategies to functionally screen soil metagenomic DNA libraries.
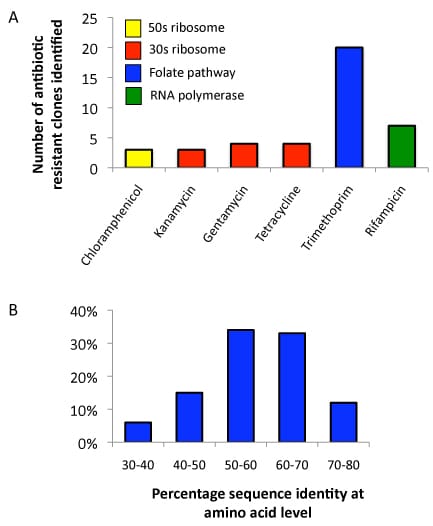
Figure 2. Antibiotic resistance profiling of a soil metagenomic library. A. Number of resistant clones recovered against each antibiotic. A library of 1.4e06 clones with an average insert size of 1.5 kb was screened against 6 antibiotics. A total of 41 resistant clones have been identified. B. Distribution of amino acid identities for 41 resistance genes recovered from soil samples compared to the most similar gene from any organism in GenBank.
Published Results
McGarvey KM, Queitsch K, and Fields S. Wide variation in antibiotic resistance proteins identified by functional metagenomic screening of a soil DNA library. Appl Environ Microbiol. 2012 Mar;78(6):1708-14. Epub 2012 Jan 13.
Download PDF
Published Results
McGarvey KM, Queitsch K, and Fields S. Wide variation in antibiotic resistance proteins identified by functional metagenomic screening of a soil DNA library. Appl Environ Microbiol. 2012 Mar;78(6):1708-14. Epub 2012 Jan 13.
Download PDF
Functional chromosomal interactions
Kevin Schutz (former lab member)
The topologies and spatial relationships of eukaryotic chromosomes are poorly understood. Together with the labs of Tony Blau, Bill Noble and Jay Shendure at the University of Washington, we developed a high-throughput method to globally capture intra- and inter-chromosomal interactions, and applied it to generate a map at kilobase resolution of the haploid genome of the budding yeast Saccharomyces cerevisiae. The map recapitulates known features of genome organization, thereby validating the method, and identifies new features. Extensive regional and higher order folding of individual chromosomes is observed. Chromosome XII exhibits a striking conformation that implicates the nucleolus as a formidable barrier to interaction between DNA sequences at either end. Inter-chromosomal contacts are anchored by centromeres and include interactions among tRNA genes, among origins of early DNA replication and among sites where chromosomal breakpoints occur. Finally, we constructed a three-dimensional model of the yeast genome. Our findings provide a glimpse of the interface between the form and function of a eukaryotic genome.
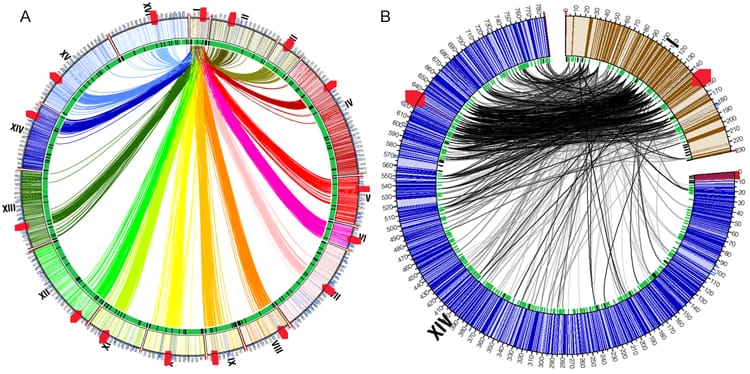
Figure 1. Inter-chromosomal interactions. A, Circos diagram showing interactions between chromosome I and the remaining chromosomes. All 16 yeast chromosomes are aligned circumferentially, and arcs depict distinct inter-chromosomal interactions. Bold red hatch marks correspond to centromeres. B, Circos diagram, generated using the intra-chromosomal interactions depicting the distinct interactions between a small and a large chromosome (I and XIV, respectively). Most of the interactions between these two chromosomes primarily involve the entirety of chromosome I, and a distinct region of corresponding size on chromosome XIV.
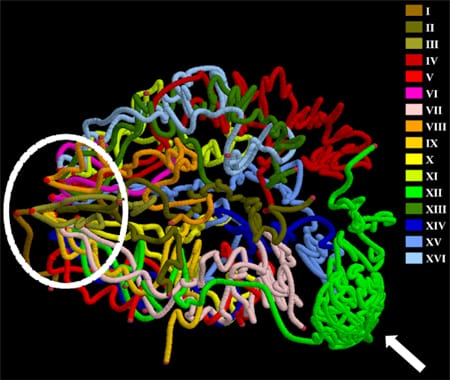
Figure 2. Three-dimensional model of the yeast genome. Chromosomes are colored individually. Centromeres and telomeres are marked by lighter and darker red dots, respectively. All chromosomes cluster via centromeres at one pole of the nucleus (the area within the dashed oval), while chromosome XII extends outward toward the nucleolus, which is occupied by rDNA repeats (indicated by the white arrow). After exiting the nucleolus, the remainder of chromosome XII interacts with the long arm of chromosome IV.
Published Results
Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA, Noble WS. A three-dimensional model of the yeast genome. Nature. 2010 May 20;465(7296):363-7.
Download PDF
Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA, Noble WS. A three-dimensional model of the yeast genome. Nature. 2010 May 20;465(7296):363-7.
Download PDF
RNA Technology
Linking barcodes via a self-splicing intro
Taylor Wang and Matt Rich (former lab members)
DNA barcodes are widely used to label cells in order to link a cell's phenotype (such as fitness in a competitive assay) to its genotype. Single barcodes are easily assayable by high-throughput sequencing. There is a need, however, for techniques to assay barcode combinations (e.g., interactions between mutations in two proteins, or all-by-all protein-protein interaction screening). Methods using targeted DNA recombination have been developed to physically link barcodes either before or after phenotyping, but these are relatively low efficiency, require significant up-front strain engineering, and in some cases have been shown to induce diploidy. We are using trans-splicing ribozymes, such as the well-studied ribosomal RNA intron from Tetrahymena thermophila, to physically link multiple barcodes.
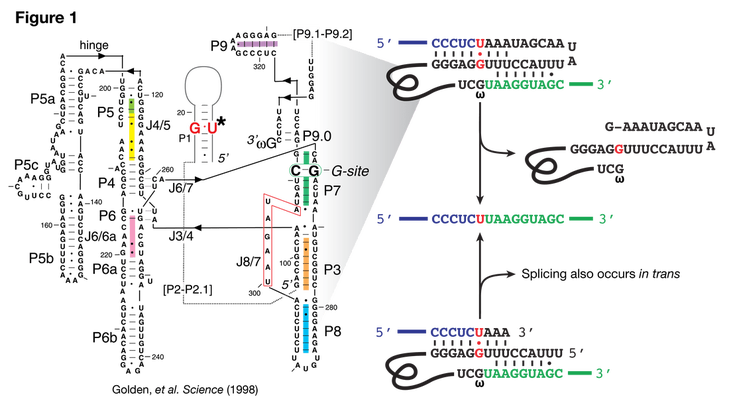
The Tetrahymena ribozyme is a self-splicing intron (Figure 1). Splicing occurs through two trans-esterification reactions targeted to a U-G wobble basepair at the 3' end of the first exon. Endogenously, the intron splices in cis (Figure 1, right), but has been shown to splice in trans both in vitro and in vivo, albeit with lower efficiency.
The Tetrahymena ribozyme is a self-splicing intron (Figure 1). Splicing occurs through two trans-esterification reactions targeted to a U-G wobble basepair at the 3' end of the first exon. Endogenously, the intron splices in cis (Figure 1, right), but has been shown to splice in trans both in vitro and in vivo, albeit with lower efficiency.
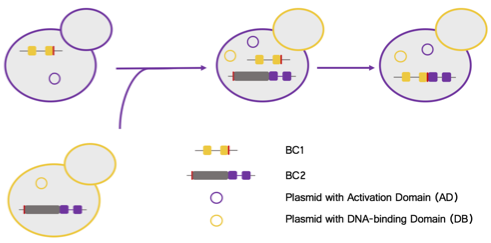
We are developing a method to use the Tetrahymena intron to physically link barcode sequences in a single RNA molecule, which can be easily assayed by high-throughput RNA sequencing. This method is being initially tested in a small library yeast two-hybrid assay, as yeast mating allows us to easily create large pools of combinatorially-barcoded strains (Figure 2).
A proof-of-principle RT-qPCR result for an in vivo trans-splicing barcode pair is shown in Figure 3A. The yeast two-hybrid signal for the nine-member array has been confirmed (Figure 3B), and the spliced barcodes of the pooled library experiment have been successfully amplified (Figure 3C). This amplification step adds Illumina adaptors and indices necessary for the final sequencing step—which is currently in progress. We plan to apply this methodology to high throughput, single pool, genetic interaction analysis, as well as all-by-all deep mutational scanning assays
Assessing the impact of synonymous mutations in TP53 and JAK3
Matt Rich, Geetha Bhagavatula, and Dave Young (former lab members)
The increased availability of DNA sequences from cancer genomes has led to the identification of mutations in oncogenes and tumor suppressor genes associated with cancer development. Traditionally, the focus has been on identifying mutations within these genes that alter the protein coding sequence, known as non-synonymous mutations. However, recent evidence has demonstrated that synonymous mutations, those in the coding regions that do not alter the protein sequence, are implicated in the development of various human diseases. Further, several oncogenes and tumor suppressor genes isolated from cancer genomes have been noted to contain more synonymous mutations than would be expected, indicating a possible role of synonymous mutations acting as driver mutations for the development of cancer. Our goal is to directly assess the potential effects of synonymous mutations in oncogenes and tumor suppressor genes through a deep mutational scanning approach: creating a library of cells containing all possible synonymous mutations for one exon of a gene and assaying the library for changes in the level of protein expression.
We are specifically investigating TP53, a tumor suppressor gene associated with a multitude of somatic cancers and a hereditary cancer syndrome, and JAK3, an oncogene recently linked with development of several leukemias. We are creating a mini-gene reporter system, in which an exon of interest flanked by one intron and exon on either side is tethered to an in-frame GFP and introduced into a human cell line via plasmid. By systematically mutating the wobble position of each codon within the exon of interest, we create a library of cells containing all possible synonymous mutations.
We are specifically investigating TP53, a tumor suppressor gene associated with a multitude of somatic cancers and a hereditary cancer syndrome, and JAK3, an oncogene recently linked with development of several leukemias. We are creating a mini-gene reporter system, in which an exon of interest flanked by one intron and exon on either side is tethered to an in-frame GFP and introduced into a human cell line via plasmid. By systematically mutating the wobble position of each codon within the exon of interest, we create a library of cells containing all possible synonymous mutations.
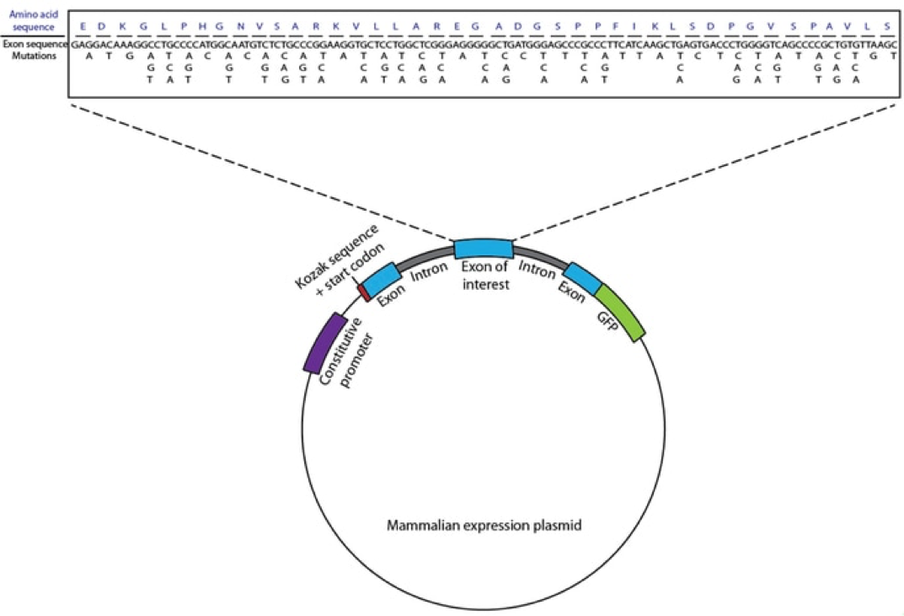
With this reporter structure, any mutations that cause changes in the level of exon expression, stability of the mRNA, splice-site skipping, or splice-site enhancement are reflected by changes in GFP level within cells. Therefore, we can use Fluorescence Activated Cell Sorting (FACS) to sort the library of cells into bins of varying fluorescence depending on the level of GFP in each cell. By high-throughput sequencing of sorted cells, we are able to determine the effect of each synonymous mutation on protein levels in the cell. Identified mutations can then be further investigated individually to characterize the possible mechanisms by which they act. This technique allows us to make more direct approximations of how much of an effect synonymous mutations can have within these genes, and expand upon the limited understanding of the potential for synonymous mutations to act as drivers for cancer development.
Published Results
Bhagavatula G, Rich MS, Young DL, Marin M, Fields S. A Massively Parallel Fluorescence Assay to Characterize the Effects of Synonymous Mutations on TP53 Expression. Mol Cancer Res. 2017 Oct;15(10):1301-1307. doi: 10.1158/1541-7786.MCR-17-0245. Epub 2017 Jun 26. PubMed PMID: 28652265; PubMed Central PMCID: PMC5626615.
Download PDF
Published Results
Bhagavatula G, Rich MS, Young DL, Marin M, Fields S. A Massively Parallel Fluorescence Assay to Characterize the Effects of Synonymous Mutations on TP53 Expression. Mol Cancer Res. 2017 Oct;15(10):1301-1307. doi: 10.1158/1541-7786.MCR-17-0245. Epub 2017 Jun 26. PubMed PMID: 28652265; PubMed Central PMCID: PMC5626615.
Download PDF
Elucidating a code for RNA sequence recognition
Daniel Melamed and Christina Miller (former lab members)
The ability to design a protein that can bind specifically to any RNA and regulate its fate would enable numerous research and therapeutic applications. However, decoding RNA-binding specificity for most types of RNA-binding domains is challenging, as these domains associate with RNA via complex networks of interactions. The challenge of engineering a domain with high specificity and affinity is further complicated by the typically weak associations these RNA-binding domains make with only a small number of RNA bases.
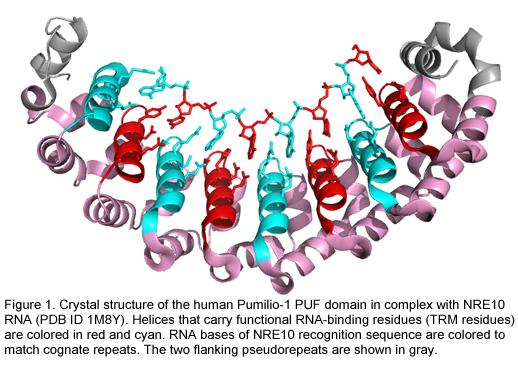
However, a few RNA-binding domains bind by recognition mechanisms that make them more ideal candidates for protein design. The modular architecture of the PUF RNA-binding domain is one such example (Figure 1). The PUF domain almost always contains 8 functional copies of a 36 amino acid long alpha-helical repeat. Each PUF repeat recognizes a single RNA base via three amino acids at conserved locations referred to here as a tripartite recognition motif (TRM) (Campbell ZT et al. 2014).
However, a few RNA-binding domains bind by recognition mechanisms that make them more ideal candidates for protein design. The modular architecture of the PUF RNA-binding domain is one such example (Figure 1). The PUF domain almost always contains 8 functional copies of a 36 amino acid long alpha-helical repeat. Each PUF repeat recognizes a single RNA base via three amino acids at conserved locations referred to here as a tripartite recognition motif (TRM) (Campbell ZT et al. 2014).
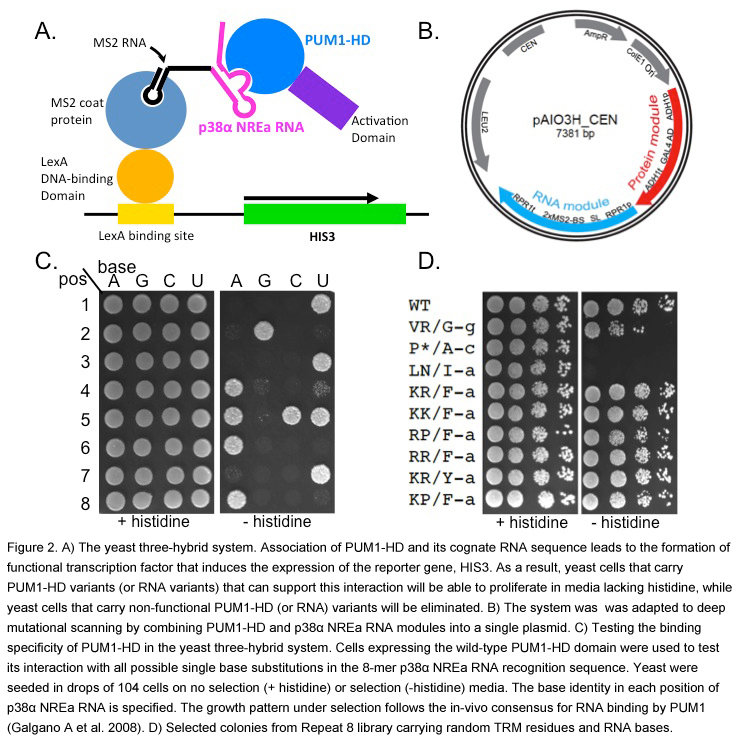
With its natural ability to bind selectively and with high affinity to its 8-mer recognition sequence, the PUF domain is an attractive candidate for deep mutational scanning, in which we elucidate the RNA-binding preference of a large number of variants. We can combine the yeast three-hybrid method (Figure 2A) with next generation sequencing technology to score the binding activities of variants of a PUF domain and a cognate RNA sequence. We adapted the traditional yeast-three hybrid expression system to deep mutational scanning by combining both the tested PUF protein and the RNA expression modules into a single, centromeric plasmid (Figure 2B). We showed that binding the specificity of each PUF repeat in this system recapitulates the in vivo specificity of this domain, and therefore we can score each mutated PUF repeat for its ability to bind to each of the 4 possible RNA bases (Figure 2C). Indeed, selection of libraries containing variants of the PUF domain and the RNA on plates that require HIS3 reporter gene activation identified TRM:RNA base combinations that are likely to interact (Figure 2D). This approach will allow us to characterize the specificity and the affinity of each PUF repeat for any RNA base and to progress towards uncovering a complete code for RNA recognition.
Codon context and translation efficiency in a yeast GFP assay
Caitlin Gamble (former lab member)
Because of degeneracy in the genetic code, several codons can encode the same amino acid. Yet variation in a gene’s synonymous codon usage can result in protein production differences with phenotypic consequences for the cell. The contexts and mechanisms by which codon usage impacts translation are not well defined.
In collaboration with the Grayhack lab at University of Rochester, we have sought to experimentally identify non-optimal codons and codon combinations in yeast. We generated two integrated libraries, each containing a three-codon insertion near the N-terminus of superfolder GFP. We performed fluorescence-activated cell sorting followed by high-throughput sequencing of the insertions to estimate the mean expression level for a total of 35,811 GFP variants. We identified a subset of codons that was frequently found in low expression variants. We also found that for a small number of adjacent codon pairs, most variants containing these pairs had low expression levels, whereas most other variants exhibited high levels of expression. Reconstructed variants with these pairs had reduced GFP fluorescence levels relative to a synonymous construct. Overall, we identified 20 pairs with evidence of a general inhibitory impact. Additionally, the directionality of a pair was often central to inhibitory effects. Thus, we have identified codon pairs that are likely to reduce translation efficiency due to the pair’s impact on translation dynamics within the ribosome, and we are currently following-up on these pairs with tRNA suppression experiments to better understand the mechanisms of codon pair-mediated inhibition.
In collaboration with the Grayhack lab at University of Rochester, we have sought to experimentally identify non-optimal codons and codon combinations in yeast. We generated two integrated libraries, each containing a three-codon insertion near the N-terminus of superfolder GFP. We performed fluorescence-activated cell sorting followed by high-throughput sequencing of the insertions to estimate the mean expression level for a total of 35,811 GFP variants. We identified a subset of codons that was frequently found in low expression variants. We also found that for a small number of adjacent codon pairs, most variants containing these pairs had low expression levels, whereas most other variants exhibited high levels of expression. Reconstructed variants with these pairs had reduced GFP fluorescence levels relative to a synonymous construct. Overall, we identified 20 pairs with evidence of a general inhibitory impact. Additionally, the directionality of a pair was often central to inhibitory effects. Thus, we have identified codon pairs that are likely to reduce translation efficiency due to the pair’s impact on translation dynamics within the ribosome, and we are currently following-up on these pairs with tRNA suppression experiments to better understand the mechanisms of codon pair-mediated inhibition.
Deep mutational scanning of a tRNA
David Young (former lab member)
tRNAs are of fundamental importance in translating the information contained in our genes into cellular and organismal function. A given tRNA must adopt a specific and conserved 3-dimensional structure in order to interact with the ribosome, with elongation factors, and with its corresponding amino acid tRNA synthetase. A good deal of cellular energy is also devoted to extensively modifying the bases of a tRNA during its maturation. Despite these constraints on tRNA shape and sequence, there are about 500 different human tRNAs and about 275 different yeast tRNAs, and significant sequence diversity both within and between species. In order to generate a set of all functional variants of a single tRNA and thereby determine the extent to which it can tolerate mutation, we have collaborated with the Phizicky and Matthews labs at the University of Rochester to adapt deep mutational scanning to the study of tRNA function.
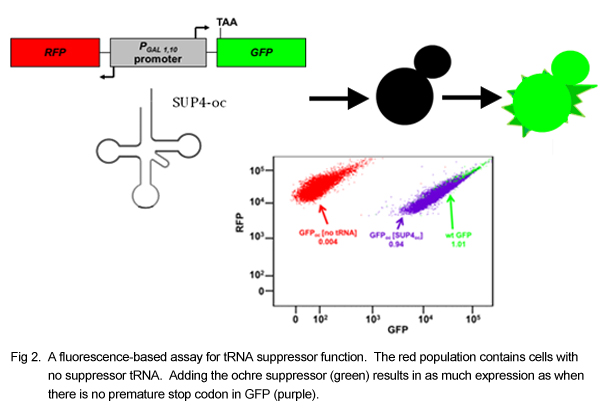
The assay relies on the ability of a suppressor tRNA, which recognizes a stop codon, to allow the ribosome to “read through” the stop codon on an mRNA instead of stopping translation at the stop codon and releasing the mRNA. We modified yeast by the addition of two plasmids, one containing a Green Fluorescent Protein (GFP) reporter and one containing a mutant version of the tyrosine tRNA that recognizes the ochre stop codon (UAA). The GFP reporter contains an ochre stop codon at the beginning of its sequence, such that it fails to be translated into a functioning protein unless a working tRNA suppressor gene is also present to read through the stop codon. In this way, the function of the ochre suppressor tRNA can be determined by observing the fluorescence of the cell. The ochre suppressor tRNA was mutated extensively, and the library of mutants was transformed into a yeast strain containing the GFP reporter. The yeast transformants were sorted by fluorescence into bins in a Fluorescence Activated Cell Sorter, and the plasmids from the yeast in each bin were sequenced on an Illumina MiSeq. For a given mutant, the percentage of MiSeq reads in each bin, along with the average fluorescence of the bins, can be used to determine the average fluorescence due to that mutant tRNA. The weighted average fluorescence was used to stratify the mutants by function.
We have obtained functional data for every possible single mutation, for about 14,000 double mutations, and for about 30,000 more highly mutated variants. Surprisingly, 37% of the single mutants retained at least some function. In addition, around 10% of double mutants showed near wild type levels of fluorescence, indicating that despite all of the modifications and structure constraints, tRNA function is relatively robust to mutation. We have also examined mutant performance in a yeast strain with a mutated Rapid tRNA Decay (RTD) quality control pathway, which degrades misfolded or unmodified tRNA. By comparing the performance of tRNA variants in the wild type strain and the decay pathway mutant, we have identified many new targets of this pathway. The majority of these new decay pathway targets are located in parts of the tRNA not previously known to be monitored by the RTD system, such as the anticodon and D stems. By examining the double mutants in relation to their constituent singles, we were able to gain some insight into the relationships between positions within the tRNA. We have seen some expected relationships; for example, deleterious mutations that abolish base pairing in one of the stems are rescued by changes that restore base pairing. Other interactions occur within or between loops. In at least one case, the variable loop can shorten to restore base pairing at the beginning of the anticodon stem. We have also tested the library at various temperatures to identify heat sensitive tRNA variants and to determine the relationship between temperature sensitivity and RTD targeting. By modeling RTD and temperature sensitivity as functions of tRNA structure and free energy, we are able to predict these values for untested tRNA mutants. We are currently validating these predictions for different mutants in different tRNAs. By examining positional interactions in various backgrounds, we hope to gain a greater understanding of the determinants of tRNA structure and function. In addition, the application of this assay to other tRNAs will provide a high throughput means of identifying potential disease causing variants.
In vivo deep mutational scanning of an RNA-recognition motif (RRM)
Daniel Melamed, David Young, and Christina Miller (former lab members)
Throughout its life, an RNA molecule associates with diverse RNA-binding proteins that regulate its processing and function. A single RNA-binding protein typically recognizes a particular subset of RNA molecules and affects their collective fate by regulating one or more steps in RNA metabolism, from pre-mRNA splicing to mRNA localization, translation and decay. Since these functions underlie multiple fundamental cellular processes, genetic changes that disrupt RNA-binding protein function can lead to multifaceted human pathologies. We are using deep mutational scanning, an experimental strategy that couples high throughput DNA sequencing with assays of protein function, to study the effects of sequence variations on the function of a common RNA-binding domain called the RNA Recognition Motif (RRM). Specifically, we made use of the necessity of a functional poly(A)-binding protein (Pab1) for yeast growth and survival to test the in vivo effects of numerous mutations in the Pab1 RRM2 domain (Figure 1). In this system, the endogenous yeast PAB1 gene has been deleted and replaced with a plasmid expressing the wild-type Pab1 from a tetracycline-regulated promoter. A second plasmid in these cells expresses one of many variants carrying random mutations in the Pab1 RRM2. Adding a tetracycline analog to the culture shuts off the expression of the wild-type gene, making the cells completely reliant on the mutant Pab1 performance for growth. High throughput sequencing of the library variants before and after addition of the tetracycline analog allows us to measure the change in frequency of each variant, which in turn can be used as a proxy for the function of the mutant Pab1 RRM domain.
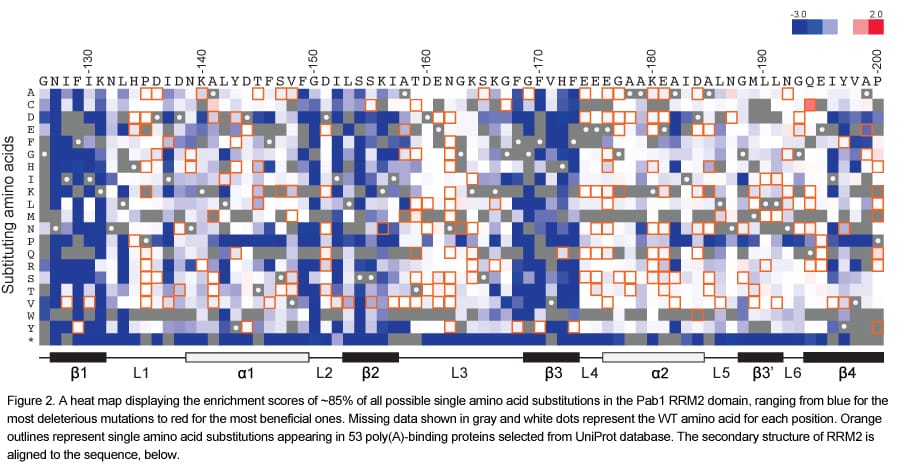
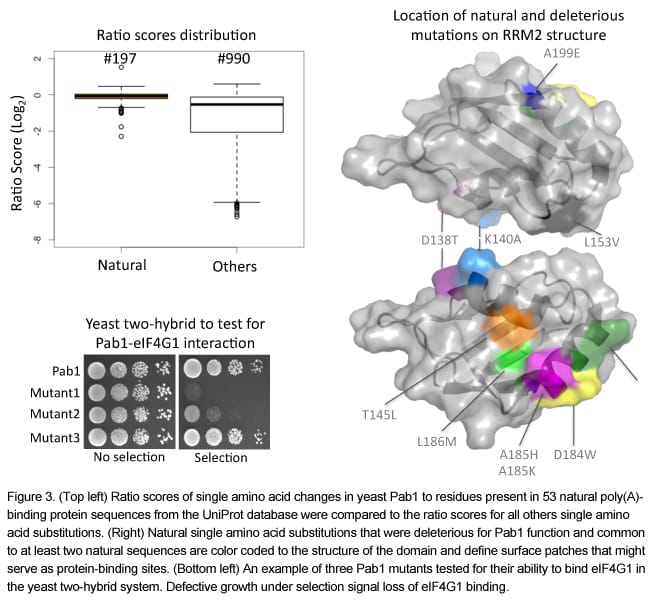
One of the major outputs of this experiment is a single amino acid substitution matrix representing all possible 19 single amino acid substitutions at each residue in the RRM2 domain of Pab1 (Figure 2). This matrix points to the β-strands as the most important for the in vivo function of RRM2, which agrees with their essential role in poly(A) binding.
To gain a better understanding of Pab1 RRM2 function, we observed the ratio scores of about 200 single amino acid substitutions that occur in other Pab1 homologous sequences (Figure 3). These scores, which can be viewed also as the output of a large-scale inter-species complementation assay, revealed that while most of the natural changes were neutral in their effects, a few substitutions were deleterious. Mapping these mutations on the RRM2 structure revealed that most of them affect residues at the protein surface. We suspected that this approach allowed the identification of protein interaction sites that diverged throughout evolution. Indeed, we found that about half of these mutations interfere with the interaction between Pab1 and the translation initiation factor eIF4G.
Overall, we suggest that extracting functional scores of naturally occurring substitutions from deep mutational scanning experiments can facilitate the identification of surface residues that were likely to co-evolve with their binding partner.
Published Results
Melamed D, Young DL, Gamble CE, Miller CR, Fields S. Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)-binding protein. RNA. 2013 Nov;19(11):1537-51. Epub 2013 Sep 24.
Download PDF
Published Results
Melamed D, Young DL, Gamble CE, Miller CR, Fields S. Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)-binding protein. RNA. 2013 Nov;19(11):1537-51. Epub 2013 Sep 24.
Download PDF
Genome-wide analysis of nascent transcription in Saccharomyces cerevisiae
Anastasia McKinlay and Carlos Araya (former lab members)
Most studies of eukaryotic gene regulation have examined mature, steady-state mRNA levels. However, steady-state mRNA levels result from the action of two opposing processes: RNA synthesis and RNA degradation. An accurate assessment of RNA synthesis is important for understanding the mechanisms that regulate gene expression.
The nuclear run-on (NRO) assay is the traditional method to directly measure RNA synthesis. We have combined the in vivo RNA labeling of this assay with high throughput DNA sequencing to examine RNA polymerase activity genome-wide in exponentially growing yeast. In parallel, we sequenced total RNA to monitor transcript abundance and compare nascent transcript and steady-state transcript levels (Figure 1A).
To analyze RNA polymerase activity within genes, we examined read density along transcribed regions. We find that in contrast to total RNA libraries, NRO libraries show a high density of reads near the 5’ ends of the transcript models, with a peak ~50 bp downstream of the transcription start site (TSS) (Figure 1B), as has been observed in human and Drosophila cells. This peak in read depth near TSSs likely indicates a promoter-proximal accumulation of paused RNA polymerase, suggesting that pausing plays a significant role in the regulation of yeast transcription. Analysis of expression levels allows us to classify genes into four classes by their activity and pausing (Figure 1C). Ranking genes by the significance of pausing reveals that histone genes are among the 5% most paused genes, suggesting that transition to productive elongation is necessary for rapid induction of histone synthesis in S phase. By calculating the ratio of NRO transcription to total RNA for each gene, we can estimate nascent transcript stabilities. This analysis has revealed that the most stable and unstable transcripts encode proteins whose functional roles are consistent with these stabilities.
Parallel analysis of nascent transcripts and steady-state transcripts with high throughput sequencing allows a genome-wide assessment of RNA polymerase activity in yeast, identifying regulatory steps of RNA synthesis and inference of RNA stabilities. We anticipate that this approach will be useful to measure changes that occur in transcription in response to environmental or genetic perturbations.
The nuclear run-on (NRO) assay is the traditional method to directly measure RNA synthesis. We have combined the in vivo RNA labeling of this assay with high throughput DNA sequencing to examine RNA polymerase activity genome-wide in exponentially growing yeast. In parallel, we sequenced total RNA to monitor transcript abundance and compare nascent transcript and steady-state transcript levels (Figure 1A).
To analyze RNA polymerase activity within genes, we examined read density along transcribed regions. We find that in contrast to total RNA libraries, NRO libraries show a high density of reads near the 5’ ends of the transcript models, with a peak ~50 bp downstream of the transcription start site (TSS) (Figure 1B), as has been observed in human and Drosophila cells. This peak in read depth near TSSs likely indicates a promoter-proximal accumulation of paused RNA polymerase, suggesting that pausing plays a significant role in the regulation of yeast transcription. Analysis of expression levels allows us to classify genes into four classes by their activity and pausing (Figure 1C). Ranking genes by the significance of pausing reveals that histone genes are among the 5% most paused genes, suggesting that transition to productive elongation is necessary for rapid induction of histone synthesis in S phase. By calculating the ratio of NRO transcription to total RNA for each gene, we can estimate nascent transcript stabilities. This analysis has revealed that the most stable and unstable transcripts encode proteins whose functional roles are consistent with these stabilities.
Parallel analysis of nascent transcripts and steady-state transcripts with high throughput sequencing allows a genome-wide assessment of RNA polymerase activity in yeast, identifying regulatory steps of RNA synthesis and inference of RNA stabilities. We anticipate that this approach will be useful to measure changes that occur in transcription in response to environmental or genetic perturbations.
Published Results
McKinlay, A., Araya, C.L. and Fields, S. Genome-wide analysis of nascent transcription in Saccharomyces cerevisiae. 2011 G3: Genes, Genomes, Genetics Dec;1(7):549-58. Epub 2011 Dec 1.
Download PDF
McKinlay, A., Araya, C.L. and Fields, S. Genome-wide analysis of nascent transcription in Saccharomyces cerevisiae. 2011 G3: Genes, Genomes, Genetics Dec;1(7):549-58. Epub 2011 Dec 1.
Download PDF
Capture and sequence analysis of RNAs containing 3' cyclic phosphate termini
Kevin Schutz (former lab member)
Standard techniques used to isolate and identify RNA from cellular extracts have traditionally relied upon hybridization to oligo-dT or T4 RNA ligase-based methodologies. These methods have been successful in isolating populations of RNAs that are modified with poly-adenosine tracts or have hydroxyl moieties (-OH) at their 3’ terminus. It is possible that these two classes represent the majority of the cellular ‘RNA universe.’ However, with the development of advanced sequencing technologies, it is also clear that the RNA universe is more complex than previously appreciated. Therefore, there is a need to develop new technologies to further profile this complexity.
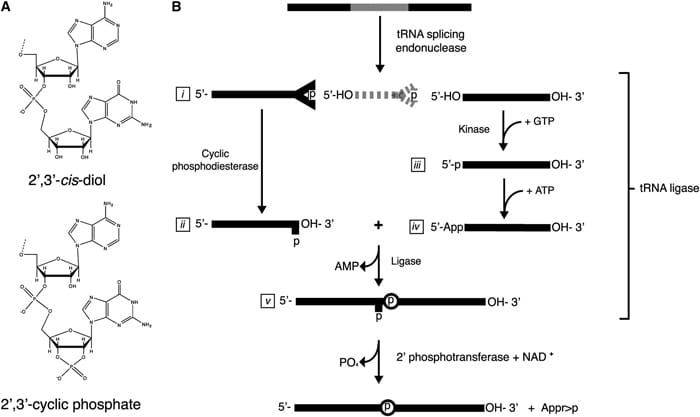
With this in mind we developed a technology that is capable of specifically isolating 2’,3’ cyclic phosphate-terminated RNAs from complex RNA mixtures. RNAs with these termini are generated as the product of particular RNA endonucleases or during ribonucleolytic cleavage. This technology uses the Arabidopsis thaliana tRNA ligase to add an adaptor oligonucleotide to RNAs that terminate in 2’,3’ cyclic phosphates. The adaptor allows specific priming by reverse transcriptase, which is followed by additional steps for PCR amplification and high throughput DNA sequencing. This method may identify processing events previously undetected by other RNA cloning techniques.
With this in mind we developed a technology that is capable of specifically isolating 2’,3’ cyclic phosphate-terminated RNAs from complex RNA mixtures. RNAs with these termini are generated as the product of particular RNA endonucleases or during ribonucleolytic cleavage. This technology uses the Arabidopsis thaliana tRNA ligase to add an adaptor oligonucleotide to RNAs that terminate in 2’,3’ cyclic phosphates. The adaptor allows specific priming by reverse transcriptase, which is followed by additional steps for PCR amplification and high throughput DNA sequencing. This method may identify processing events previously undetected by other RNA cloning techniques.
Published Results
Schutz K, Hesselberth JR, Fields S. Capture and sequence analysis of RNAs with terminal 2',3'-cyclic phosphates. RNA. 2010 Mar;16(3):621-31.
Download PDF
Supplemental Figures
Supplemental Figure Legends
Supplemental Raw Sequencing Data
Schutz K, Hesselberth JR, Fields S. Capture and sequence analysis of RNAs with terminal 2',3'-cyclic phosphates. RNA. 2010 Mar;16(3):621-31.
Download PDF
Supplemental Figures
Supplemental Figure Legends
Supplemental Raw Sequencing Data
Protein Technology
Many of the projects described below rely on a method developed in our lab called Deep Mutational Scanning (DMS).
Published paper on Deep Mutational Scanning:
Fowler DM, Fields S. Deep mutational scanning: a new style of protein science. Nature Methods. 2014 Aug;11(8):801-7.
Published paper on Deep Mutational Scanning:
Fowler DM, Fields S. Deep mutational scanning: a new style of protein science. Nature Methods. 2014 Aug;11(8):801-7.
Polypeptide inhibitors of Src kinase
Mike Dorrity and Ben Brandsen (former lab member)
Dominant negative polypeptides can act as inhibitors by binding to the wild type protein or by titrating an essential ligand. In nature, they frequently arise from truncated fragments of a wild type protein. Given this origin of natural dominant negatives, one rich source for finding dominant negatives in the laboratory might be short fragments of a protein that occupy key binding sites on the full-length protein. Additionally, since many proteins contain intramolecular regulatory domains, short protein fragments that relieve autoinhibitory function and lead to enhanced activation of the wild type protein might be identified as dominant negatives.
Using the inherent toxicity of human Src protein kinase expression in yeast, we screened tens of thousands of Src fragments for their capacity to relieve Src toxicity (Fig. 1a). By deep sequencing the fragments before and after selection, we identify fragments that are either enriched or depleted during selection (Fig 1b). Enriched fragments represent potential inhibitors of Src, while depleted fragments represent potential activators of Src. The most enriched and most depleted fragments are being assessed for their ability to inhibit or activate the phosphotransferase activity of Src kinase. We hope to use this method to identify variant-specific inhibitors of Src and other human protein kinases that might be used as therapeutics.
Using the inherent toxicity of human Src protein kinase expression in yeast, we screened tens of thousands of Src fragments for their capacity to relieve Src toxicity (Fig. 1a). By deep sequencing the fragments before and after selection, we identify fragments that are either enriched or depleted during selection (Fig 1b). Enriched fragments represent potential inhibitors of Src, while depleted fragments represent potential activators of Src. The most enriched and most depleted fragments are being assessed for their ability to inhibit or activate the phosphotransferase activity of Src kinase. We hope to use this method to identify variant-specific inhibitors of Src and other human protein kinases that might be used as therapeutics.
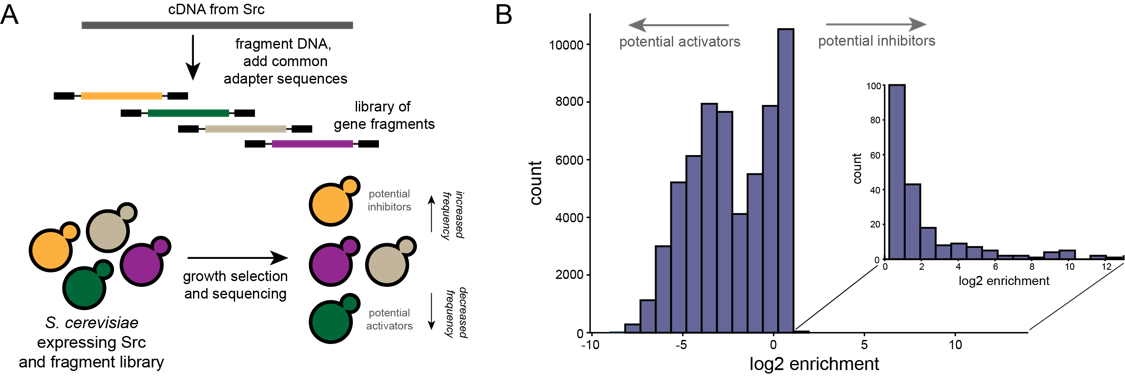
Figure 1. A. Selection strategy to find polypeptide inhibitors of Src kinase. B. Distribution of enrichment scores for fragments before and after selection.
Balance between mutually exclusive traits shifted by variants of a yeast transcription factor
Michael Dorrity (Collaboration with the Queitsch Lab, University of Washington Department of Genome Sciences)
Uncovering the genetic underpinnings of complex traits has proven difficult. From crop yield to autism, variants identified in genome-wide association studies (GWAS) explain only a small fraction of the heritable phenotypic variation, leaving a significant gap in our understanding. Using the mating pathway of Saccharomyces cerevisiae (Fig. 1A), we seek to develop a model for testing hypotheses about complex trait genetics. For example: Does most variation underlying complex traits act additively or epistatically? What proportion of mutational effects are subject to environment? Do known genetic modifiers like the chaperone Hsp90 act on this variation? We make controlled modifications to the genetic architecture of mating and examine phenotypic output to develop expectations for the translation of genotype to phenotype. To do so, we utilize deep mutational scanning, a method that links a phenotypic output to a library of genetic variants via high-throughput sequencing. This method allows us to identify small-effect mutations in individual genes, as well as combinatorial effects of many small-effect mutations across multiple genes.
Effects of mutations in individual mating pathway components (Fig. 1B) are systematically determined by introducing tens of thousands of protein variants into large populations of yeast which are then subjected to selection for mating efficiency (Fig. 1C). Furthermore, variants are tested in the absence of strong genetic modifiers like the protein chaperone Hsp90 as well as under varying stress conditions to uncover variants with genetic and environmental dependencies, respectively. After determining individual effects of very large pools of variants, we test mutant libraries for each mating gene in combination (Fig. 1D) in order to empirically determine the role of epistasis between mating genes. This design allows us to comprehensively show how additive genetic variation, epistatic interactions, and environmental factors contribute to a complex trait.
Effects of mutations in individual mating pathway components (Fig. 1B) are systematically determined by introducing tens of thousands of protein variants into large populations of yeast which are then subjected to selection for mating efficiency (Fig. 1C). Furthermore, variants are tested in the absence of strong genetic modifiers like the protein chaperone Hsp90 as well as under varying stress conditions to uncover variants with genetic and environmental dependencies, respectively. After determining individual effects of very large pools of variants, we test mutant libraries for each mating gene in combination (Fig. 1D) in order to empirically determine the role of epistasis between mating genes. This design allows us to comprehensively show how additive genetic variation, epistatic interactions, and environmental factors contribute to a complex trait.
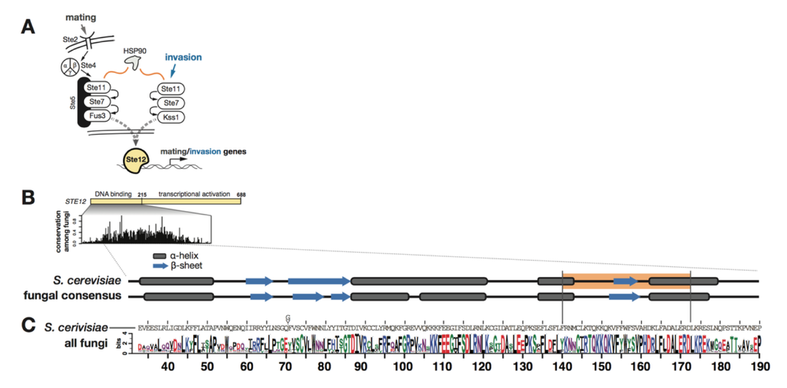
Figure 1. Conservation of Ste12 and Tec1 and their DNA-binding sites.
We have also expanded our approach to include multiple complex traits. We investigated the effect of a common set of biochemical changes in the transcription factor Ste12 (Fig. 1A, 1B, 1C) in promoting two different phenotypes in yeast: mating and invasion. In Saccharomyces cerevisiae, the decision to mate or invade relies on environmental cues that converge on a shared transcription factor, Ste12. Specificity toward invasion occurs via Ste12 binding cooperatively with the co-factor Tec1. We have characterized the in vitro binding preferences of Ste12 to identify a defined spacing and orientation of dimeric sites, one that is common in pheromone-regulated genes (Fig 2A, 2B). We find that single amino acid changes in the DNA-binding domain of Ste12 can shift the preference of yeast toward either mating or invasion (Fig. 1E).
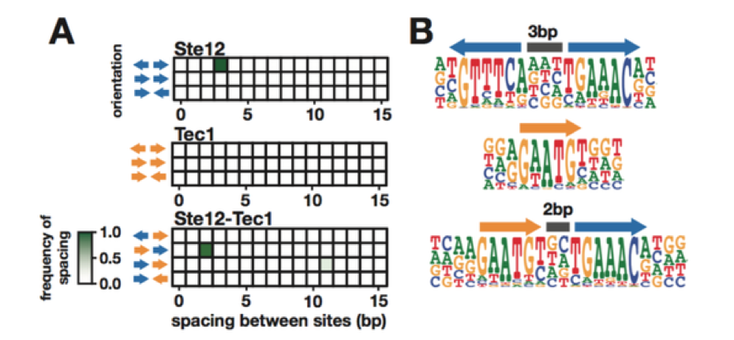
Figure 2. Identification of the DNA-binding preferences of Ste12 and Tec1 by HT-SELEX.
These mutations define two distinct regions of this domain, suggesting alternative modes of DNA binding for each trait. Some exceptional Ste12 mutants promote hyperinvasion in a Tec1-independent manner; these fail to bind cooperative sites with Tec1 and bind to unusual dimeric Ste12 sites that contain one highly degenerate half site. We propose a model for how activation of invasion genes could have evolved with Ste12 alone (Fig. 3).
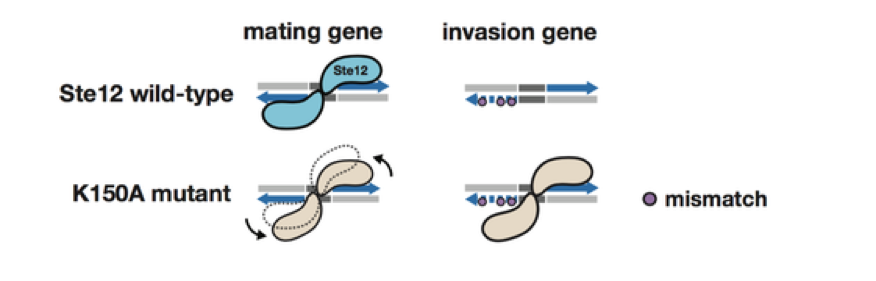
Figure 3. A model for novel site recognition by Tec1-independent Ste12 variants.
We also introduced tens of thousands of variants in three interacting mating pathway genes: the scaffold protein STE5, the MAPKKK STE11, and the MAPKK STE7. We subjected yeast expressing one of these variant libraries to a selection for mating ability, and used high-throughput sequencing to determine the mating proficiency of each variant. From these data, we identified key residues for mating that are sensitive to mutations, as well as many neutral to slightly deleterious mutations.
In future experiments, we will select several hundred small effect variants in each gene and combine these variants in two mating pathway genes at a time. We will then test these double mutants for mating ability. This experiment will allow us to measure the degree of epistasis between tens of thousands of variants in multiple interacting proteins in a single pathway.
In future experiments, we will select several hundred small effect variants in each gene and combine these variants in two mating pathway genes at a time. We will then test these double mutants for mating ability. This experiment will allow us to measure the degree of epistasis between tens of thousands of variants in multiple interacting proteins in a single pathway.
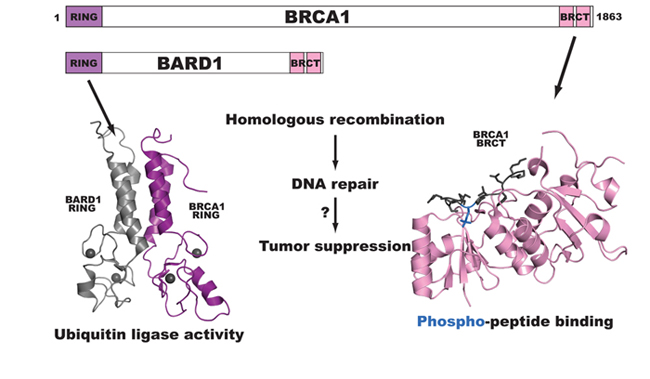
Towards testing all 35,397 possible missense variants of BRCA1 for function
Lea Starita and Justin Gullingsrud (former lab members)
BRCA1 is a breast and ovarian cancer-specific tumor suppressor gene and has been subject to much diagnostic sequencing. Multiple cancer-predisposing mutations have been identified along with >500 missense variants classified as Variants of Uncertain Significance or VUS. BRCA1 is an 1863 amino acid protein with two recognizable domains. The N-terminus contains a RING domain and is part of an active ubiquitin ligase and the C-terminus has tandem BRCT (BRCA1 C-Terminus) repeats that bind to phosphorylated peptides and activate transcription. BRCA1 is required for double-strand DNA break repair via homologous recombination, and mutations throughout the protein have deleterious effects on this function. We have devised several assays to score all of the 35,397 possible missense variants of BRCA1 for effects on the protein’s biochemical and cellular functions using a method of deep mutational scanning.
We scored 2413 of the possible 5757 missense variants (40%) of the N-terminal 304 amino acids of BRCA1 for ubiquitin ligase function using a phage display system that selects for active variants in an in vitro autoubiquitination reaction. Within these variants 57 have been identified in patients as VUS. The range of ubiquitin ligase function of the VUS variants varied from nearly completely inactive to fully functional, suggesting that some of the variants of BRCA1 that are classified as VUS are nonfunctional ubiquitin ligases.
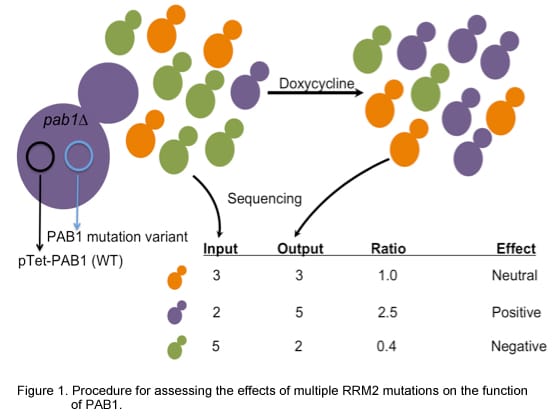
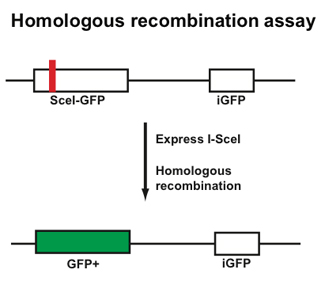
To assess the effect of mutation on ubiquitin ligase function a library of coding variants of the RING domains of BRCA1-BARD1 is fused to the T7 bacteriophage coat protein. The E3-phage are subjected to in vitro ubiquitination reactions followed by selection for phage coding for active E3 ligase (as outlined in the flow diagram below). Phages harboring active E3 ligases increase in abundance throughout selection whiles phages harboring E3 ligases with deleterious mutations decrease in abundance. These changes are measured by sequencing the input and selected populations. Enrichment ratios (E) are calculated by dividing the frequency at which each variant occurs in the selected population by its frequency in the input population.
To assess the effect of mutation on ubiquitin ligase function a library of coding variants of the RING domains of BRCA1-BARD1 is fused to the T7 bacteriophage coat protein. The E3-phage are subjected to in vitro ubiquitination reactions followed by selection for phage coding for active E3 ligase (as outlined in the flow diagram below). Phages harboring active E3 ligases increase in abundance throughout selection whiles phages harboring E3 ligases with deleterious mutations decrease in abundance. These changes are measured by sequencing the input and selected populations. Enrichment ratios (E) are calculated by dividing the frequency at which each variant occurs in the selected population by its frequency in the input population.
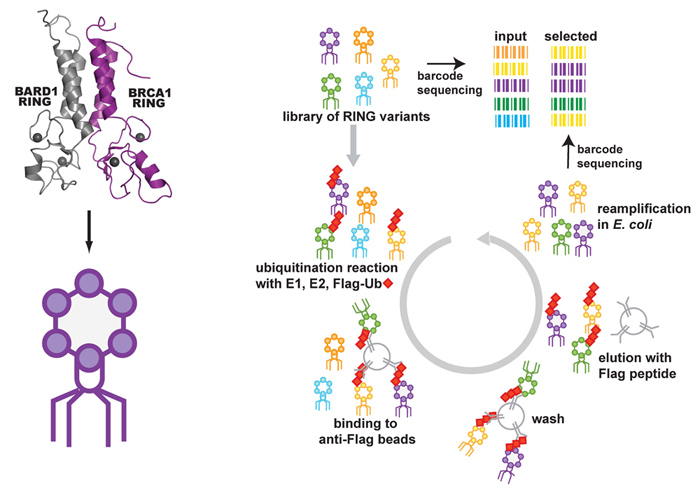
We then compared the Enrichment ratio (E) scores for each variant from the deep mutational scan of the RING domain of BRCA1 to the BRCA1 informational database classification. 2356 variants were never found in the human population and, as expected, the E scores for these variants ranged from completely inactive to highly active. 57 of the variants were classified as VUS and many of these are nonfunctional ubiquitin ligases in our assay.
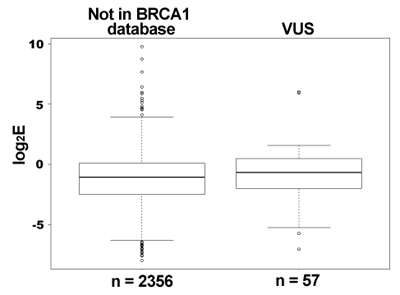
Finally, we are using a cell-based assay to score the effect of missense mutations in full-length BRCA1 on the ability of these variant BRCA1 proteins to rescue homologous recombination when the endogenous protein is repressed. To this end we are optimizing the molecular manipulations to build the libraries of variants with single amino acid changes into lentiviral vectors to transduce into a homologous recombination-reporter cell line.
Published Results
Starita LM, Young DL, Islam M, Kitzman JO, Gullingsrud J, Hause RJ, Fowler DM, Parvin JD, Shendure J, Fields S. Massively Parallel Functional Analysis of BRCA1 RING Domain Variants. Genetics. 2015 Mar 30. pii: genetics.115.175802.
Starita LM, Young DL, Islam M, Kitzman JO, Gullingsrud J, Hause RJ, Fowler DM, Parvin JD, Shendure J, Fields S. Massively Parallel Functional Analysis of BRCA1 RING Domain Variants. Genetics. 2015 Mar 30. pii: genetics.115.175802.
Uncovering the structural basis of GPCR functional selectivity through deep mutational scanning
David Young (former lab member)
G Protein Coupled Receptors (GPCRs) are a diverse family of plasma membrane bound proteins that all share 7 transmembrane helices, 3 intracellular loops, and 3 extracellular loops. There are close to 800 human GPCR genes which are responsible for a large proportion of the cellular communication in our species. Approximately 369 of these are non-sensory, making them current or potential drug targets. An estimated 40-60% of current therapeutic drugs target at least one GPCR, so advances in our understanding of signal transduction through GPCRs have potentially widespread clinical ramifications.
It has recently become clear that for any given G Protein Coupled Receptor (GPCR), multiple signaling pathways might be activated and multiple mechanisms might lead to receptor internalization at different rates depending on the specific ligand being used. This phenomenon is called “functional selectivity” or “biased agonism,” and its molecular and structural basis is only just starting to be elucidated. Though recent advances in crystallographic techniques have lead to an increasing number of structures for both active and inactive GPCRs, much remains unclear about the mechanisms of functional selectivity.
It has recently become clear that for any given G Protein Coupled Receptor (GPCR), multiple signaling pathways might be activated and multiple mechanisms might lead to receptor internalization at different rates depending on the specific ligand being used. This phenomenon is called “functional selectivity” or “biased agonism,” and its molecular and structural basis is only just starting to be elucidated. Though recent advances in crystallographic techniques have lead to an increasing number of structures for both active and inactive GPCRs, much remains unclear about the mechanisms of functional selectivity.
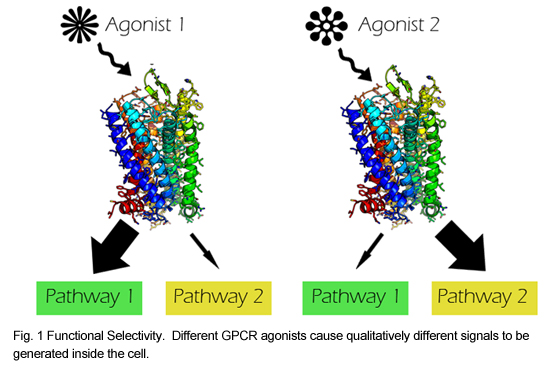
We are currently developing a set of high throughput assays for interrogating the effects of all single mutations in a GPCR on receptor expression, internalization, and signaling in a system that has already been shown to display functional selectivity: the Mu Opioid Receptor (MOR). This receptor is clinically important, as it’s the major target of opioid analgesics. Functionally distinct opioid agonists result in different amounts of tolerance development. Also, it is thought that several of the negative side effects of opioids, such as constipation and respiratory depression, might be mediated by a different pathway than their analgesic effects, which would make functional selectivity in this receptor particularly interesting clinically.
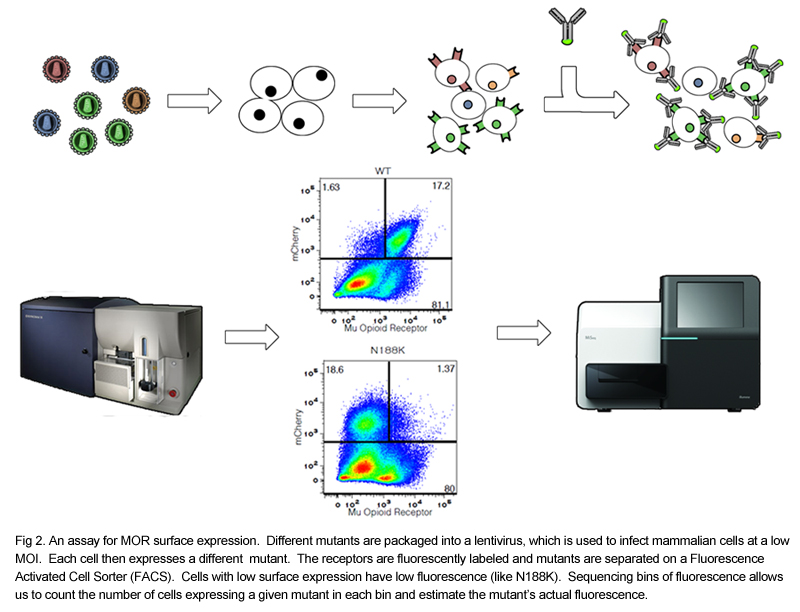
We have created a normalizable mammalian expression system for the MOR and cloned several mutants with known defects in cell surface expression into a lentiviral vector. We have demonstrated the feasibility of separating mutants based on their surface expression using a fluorescent antibody and flow cytometry. By binning cells by the amount of fluorescence, we can separate poorly expressed mutants from highly expressed mutants, and we can determine the contents of each bin by sequencing. We are currently generating a library of all single mutants of the MOR. Additional assays for receptor internalization and inhibition of calcium release will be developed after the library is created and functional selectivity will be examined by comparing results between assays using different agonists. Data generated in this project will complement structural data based on NMR and X ray crystallography by providing a functional map to overlay on the spatial one.
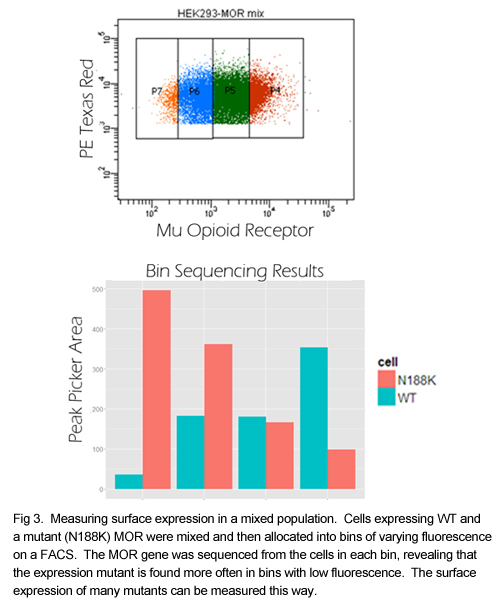
High-throughput analysis of a protein degradation signal
Griffin Kim, Christina Miller, and David Young (former lab members)
Determining the half-life of proteins is critical for an understanding of virtually all cellular processes. Current methods for measuring in vivo protein stability, including large-scale approaches, are limited in their throughput or in their ability to discriminate among small differences in stability. We developed a new method, Stable-seq, which uses a simple genetic selection combined with high-throughput DNA sequencing to assess the in vivo stability of a large number of variants of a protein. The variants are fused to a metabolic enzyme, which here is the yeast Leu2 protein. Plasmids encoding these Leu2 fusion proteins are transformed into yeast, with the resultant fusion proteins accumulating to different levels based on their stability and leading to different doubling times when the yeast are grown in the absence of leucine. Sequencing of an input population of variants of a protein and the population of variants after leucine selection allows the stability of tens of thousands of variants to be scored in parallel. By applying the Stable-seq method to variants of the protein degradation signal Deg1 from the yeast Matα2 protein, we generated a high-resolution map that reveals the effect of ~30,000 mutations on protein stability. The scores determined by Stable-seq of variants carrying single mutations are visualized in this heat map, with cell growth rates that would correspond to scores shown in the inset boxes.
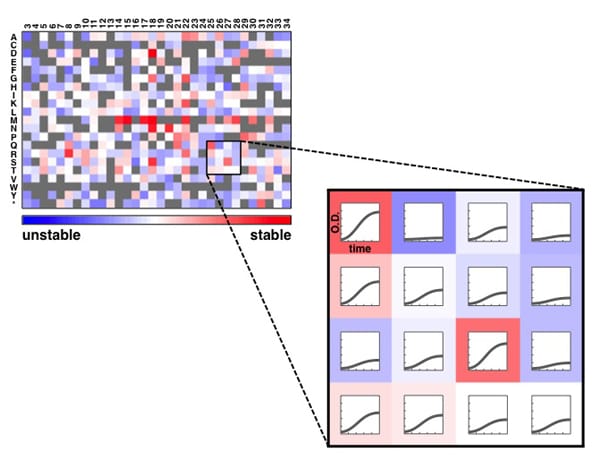
We identified mutations that likely affect stability by changing the activity of the degron, by leading to translation from new start codons, or by affecting N-terminal processing. Stable-seq should be applicable to other organisms via the use of suitable reporter proteins, as well as to the analysis of complex mixtures of fusion proteins.
Published Results
Kim I, Miller CR, Young DL, Fields S. High-throughput Analysis of in vivo Protein Stability. Mol Cell Proteomics. 2013 Jul 29.
Published Results
Kim I, Miller CR, Young DL, Fields S. High-throughput Analysis of in vivo Protein Stability. Mol Cell Proteomics. 2013 Jul 29.
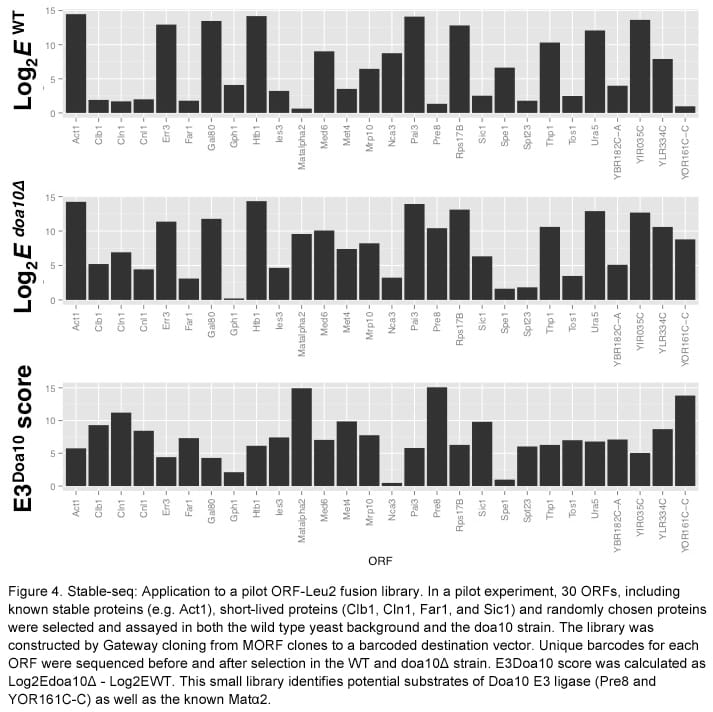
Stable-Seq: a new approach to define the specificity of E3 ligases to substrates of the ubiquitin proteasome system
Griffin Kim and Christina Miller (former lab members)
The ubiquitin proteasome system (UPS) is a complex pathway in which hundreds of regulatory proteins are involved in recognizing protein substrates, tagging them with ubiquitin, and degrading them by the proteasome. A deeper understanding of the regulatory mechanism of this system is key to developing treatments for UPS-related diseases, such as cancer and neurodegenerative disorders. A fundamental question in this field is the determination of which substrates are processed by which regulators. Among more than 100 E3 ligases in yeast, which are primarily responsible for substrate recognition, only a few substrates have been assigned to specific E3 ligases. To delineate substrate specificity of these E3 ligases, we are applying a method we have developed, called Stable-seq. In this method, we fuse either a random sequence or an open reading frame (ORF) to a nutritional marker. The random sequence or ORF determines the stability of the fusion, such that selection for the nutritional marker leads to differential growth rates. The cells grow slower in a wild type strain in which the degradation signal or ORF is unstable, but they grow faster in a strain lacking an E3 enzyme that is crucial for the degradation.
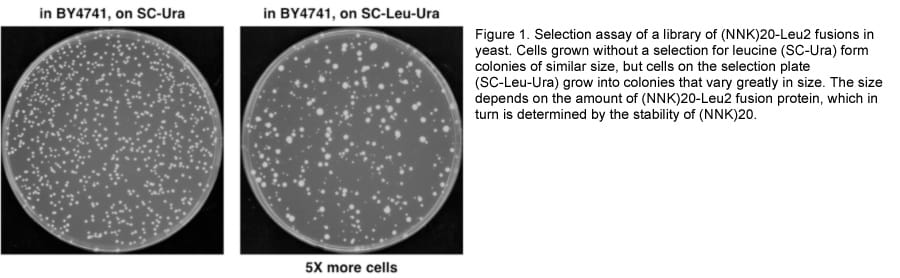
To this end, we fused random sequences (a stretch of 20 NNK codons) to the LEU2 gene and assayed by Stable-seq. The selection plate (-Leu –Ura) shows that the random sequence fusion library results in differential growth rates in the wild type strain (Figure 1), and synthetic degradation signals (synD) identified by the high-throughput sequencing and analysis from a pilot experiment have been confirmed by spotting assay (Figure 2).
To this end, we fused random sequences (a stretch of 20 NNK codons) to the LEU2 gene and assayed by Stable-seq. The selection plate (-Leu –Ura) shows that the random sequence fusion library results in differential growth rates in the wild type strain (Figure 1), and synthetic degradation signals (synD) identified by the high-throughput sequencing and analysis from a pilot experiment have been confirmed by spotting assay (Figure 2).

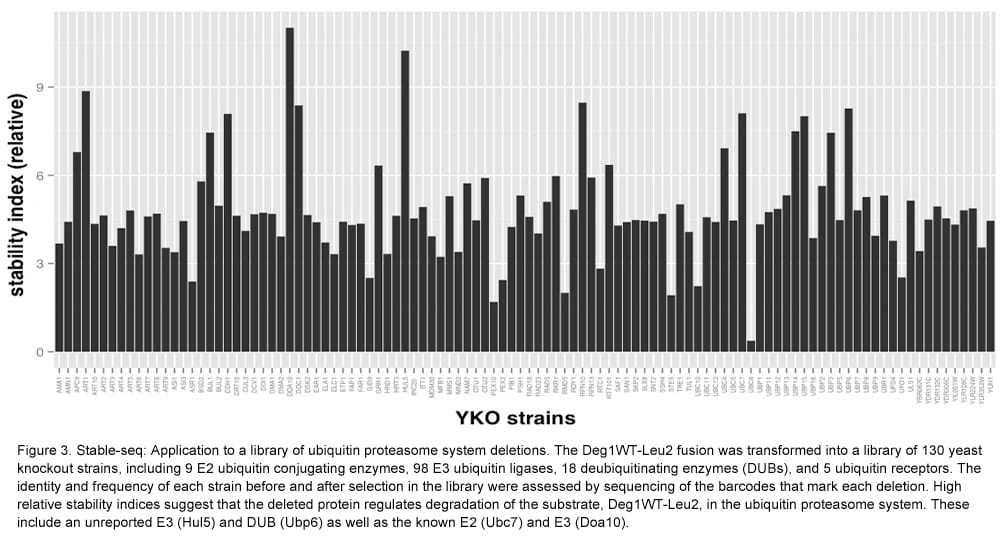
At the same time, nearly every yeast ORF was transferred from a movable-ORF (MORF) library to a destination vector, which fuses them to the LEU2 gene by Gateway cloning. To determine how well this proxy for stability works in yeast knockout (YKO) strains, we tested the Deg1-Leu2 fusion. Deg1-Leu2 fusion plasmids were transformed into a pool of 130 YKO strains. The deletion of the DOA10 gene, encoding the relevant E3 enzyme, resulted in the greatest increase in stability (Figure 3). We also tested a small library of 30 ORF-Leu2 fusions. By assaying the library in a doa10 deletion strain, we identified potential substrates whose stability increased compared to that in the wild type strain (Figure 4). Stable-seq may enable a proteome-wide effort to measure in vivo protein stability and to pair E3 enzymes with their substrates.
Deep mutational scanning to analyze protein function
Doug Fowler, Carlos Araya, and Jason Stephany (former lab members)
Understanding the functional and biophysical characteristics of proteins is of paramount importance. We have developed a method, deep mutational scanning (Figure 1), that makes use of protein display technology in conjunction with high-throughput sequencing. Deep mutational scanning enables the investigation of protein function on an unprecedented scale, facilitating the simultaneous measurement of the fitness of hundreds of thousands of mutants of a protein.
Protein display technologies physically link proteins and the DNA sequences that encode them. Protein display allows for selection among a large library of protein variants for those with a protein function. Protein display technology has been restricted in scope by the requirement for back-end DNA sequencing, which has limited the number of selected protein variants that can be identified to a few hundred. Deep mutational scanning alleviates this bottleneck by using high-throughput sequencing to sequence tens of millions of individual library members in parallel (Figure 1). The primary benefit of this approach is that millions of protein variants can be simultaneously identified and counted. Comparison of the frequency of a given variant in a selected library and in the input library yields an enrichment ratio that is an estimate of function. The key ingredients—protein display, low-intensity selection and highly accurate, high throughput sequencing—are simple and becoming widely available. Deep mutational scanning data can be used to construct protein sequence–function maps, and systematic analysis of deep mutational scanning data can reveal fundamental protein properties. We have applied deep mutational scanning to a number of proteins in a variety of functional assays.
Protein display technologies physically link proteins and the DNA sequences that encode them. Protein display allows for selection among a large library of protein variants for those with a protein function. Protein display technology has been restricted in scope by the requirement for back-end DNA sequencing, which has limited the number of selected protein variants that can be identified to a few hundred. Deep mutational scanning alleviates this bottleneck by using high-throughput sequencing to sequence tens of millions of individual library members in parallel (Figure 1). The primary benefit of this approach is that millions of protein variants can be simultaneously identified and counted. Comparison of the frequency of a given variant in a selected library and in the input library yields an enrichment ratio that is an estimate of function. The key ingredients—protein display, low-intensity selection and highly accurate, high throughput sequencing—are simple and becoming widely available. Deep mutational scanning data can be used to construct protein sequence–function maps, and systematic analysis of deep mutational scanning data can reveal fundamental protein properties. We have applied deep mutational scanning to a number of proteins in a variety of functional assays.

Published Results
Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D, Fields S. High-resolution mapping of protein sequence-function relationships. Nat Methods. 2010 Sep;7(9):741-6.
Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D, Fields S. High-resolution mapping of protein sequence-function relationships. Nat Methods. 2010 Sep;7(9):741-6.
Systematic analysis of large scale fitness data to identify mutations that stabilize proteins
Doug Fowler and Carlos Araya (former lab members)
Enhancing protein stability is often critical for industrial and pharmaceutical applications. Stabilizing mutations permit acquisition of other, destabilizing mutations that improve function. This phenomenon can be observed as epistasis, where multiple mutations combine with unpredictable fitness effects. We identify stabilizing mutations in a WW domain based solely on parallel measurement of the fitness of 47,000 variants to bind to a peptide ligand and subsequent calculation of >5,000 epistasis scores (Figure 2A). We introduce an epistasis-based metric, “partner potentiation,” which identified 15 candidate stabilizing mutations, including three known stabilizing mutations (Figure 2B). We tested six novel candidates by thermal denaturation and found two highly stabilizing mutations, one more stabilizing than any previously known mutation. Thus, systematic analysis of large-scale protein fitness data can reveal fundamental physicochemical properties such as stability.
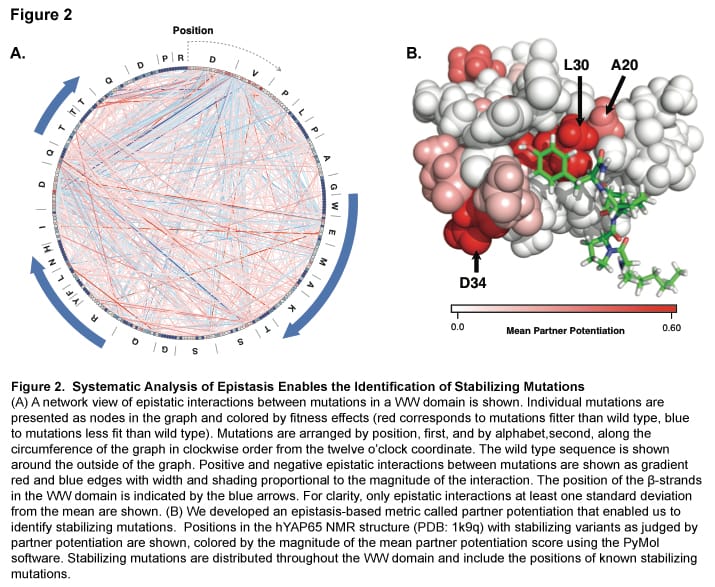
Published Results
Araya CL, Fowler DM, Chen W, Muniez I, Kelly JW, Fields S. A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc Natl Acad Sci U S A. 2012 Oct 16;109(42):16858-63.
Download PDF
Araya CL, Fowler DM, Chen W, Muniez I, Kelly JW, Fields S. A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc Natl Acad Sci U S A. 2012 Oct 16;109(42):16858-63.
Download PDF
Understanding the Molecular Basis of Selectivity in the Protein Kinase A/AKAP-79 interaction
Doug Fowler and Jason Stephany (Collaboration with the Scott Lab, HHMI and University of Washington Department of Pharmacology)
Protein Kinase A (PKA) is a central intracellular protein kinase that regulates the activity of many proteins involved in cellular metabolism. PKA activity is controlled via interactions with A Kinase Anchoring Proteins (AKAPs). AKAPs function by binding to the PKA regulatory subunit, localizing PKA within the cell. AKAPs can interact with either the alpha or the beta isoform of the regulatory subunit of PKA, or they can interact with both. The alpha and beta isoforms are highly similar, making it difficult to study the molecular determinants of selectivity between isoforms.
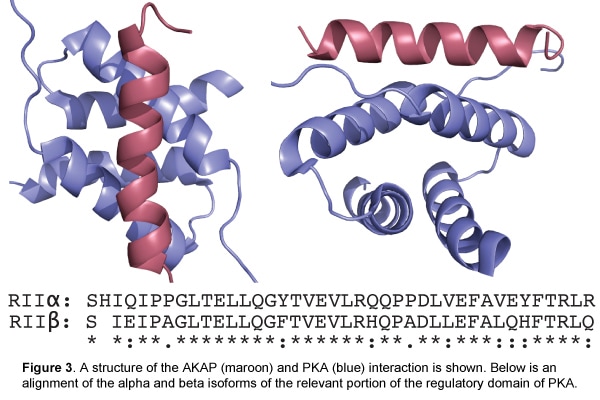
We are using phage display in combination with high-throughput sequencing to identify the sequence determinants of AKAP selectivity. We displayed a library of millions of mutagenized AKAP proteins on the surface of T7 phage and then subjected this library to selection against either the alpha or beta isoform of the regulatory subunit of PKA. By comparing the abundance of each variant before and after selection, we derived enrichment ratios for several hundred thousand variants. Most variants performed similarly in selections against both the alpha and beta isoforms. However, some variants displayed strong selectivity for either the alpha or beta isoform. We are using the results of this assay to develop highly alpha- and beta-specific AKAPs. These highly specific AKAPs will bind only to PKAs with the cognate regulatory isoform. If introduced into cells at high concentrations, they will disrupt the normal regulatory interaction for their cognate isoform, enabling us to study the biological significance of the isoforms.
Enrich: software for analysis of protein function by enrichment and depletion of variants
Doug Fowler and Carlos Araya (former lab members)
We developed Enrich, a tool for analyzing deep mutational scanning data. Enrich identifies all unique variants (mutants) of a protein in high-throughput sequencing data sets and can correct for sequencing errors using overlapping paired-end reads. Enrich uses the frequency of each variant before and after selection to calculate an enrichment ratio, which is used to estimate fitness. Enrich provides an interactive interface to guide users. It generates user-accessible output for downstream analyses as well as several visualizations of the effects of mutation on function, thereby allowing the user to rapidly quantify and comprehend sequence–function relationships. Enrich is implemented in Python, is available under a FreeBSD license and can be downloaded here. Enrich includes detailed documentation as well as a small example data set.
Published Results
Fowler DM, Araya CL, Gerard W, Fields S. Enrich: Software for Analysis of Protein Function by Enrichment and Depletion of Variants. Bioinformatics. 2011 Oct 17.
Published Results
Fowler DM, Araya CL, Gerard W, Fields S. Enrich: Software for Analysis of Protein Function by Enrichment and Depletion of Variants. Bioinformatics. 2011 Oct 17.
Activity-enhancing mutations in an E3 ubiquitin ligase discovered by deep mutational scanning
Lea Starita and Russell Lo (former lab members)
Although ubiquitination plays a critical role in virtually all cellular processes, understanding of the mechanistic details of ubiquitin transfer is still rudimentary. To identify the molecular determinants with E3 ligases that modulate activity, we developed a high-throughput assay (Figure 1) to measure the activity of nearly 100,000 protein variants of the U-box domain of murine Ube4b and found rare mutations that enhanced activity both in vitro and in cellular p53 degradation assays. Our results highlight the utility of high-throughput mutagenesis in delineating the molecular basis of enzyme activity.
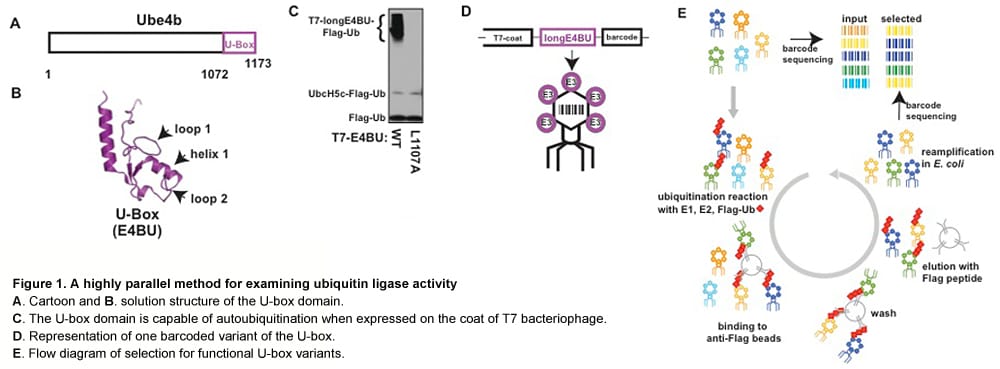
Published Results
Starita LM, Pruneda JN, Lo RS, Fowler DM, Kim HJ, Hiatt JB, Shendure J, Brzovic PS, Fields S, Klevit RE. Activity-enhancing mutations in an E3 ubiquitin ligase identified by high-throughput mutagenesis. Proc Natl Acad Sci U S A. 2013 Mar 18.
Download PDF
Starita LM, Pruneda JN, Lo RS, Fowler DM, Kim HJ, Hiatt JB, Shendure J, Brzovic PS, Fields S, Klevit RE. Activity-enhancing mutations in an E3 ubiquitin ligase identified by high-throughput mutagenesis. Proc Natl Acad Sci U S A. 2013 Mar 18.
Download PDF
Investigating the HIV-1 Tat-TAR interaction
Daniel Melamed, Matt Rich, and Christina Miller (former lab members)
The HIV-1 Tat protein is integral to the viral life-cycle as it can induce efficient transcription of the virus after binding a folded element of the HIV LTR called TAR. Previous studies have elucidated the effects of some mutations of Tat, but the overall depth and density of the studied mutations is low. We are investigating the Tat-TAR interaction using deep mutational scanning, a high-throughput technology recently developed in the lab.
By creating a library of hundreds of thousands of variants of Tat and selecting for binding to TAR using a yeast three-hybrid assay, we are examining the relationship between the sequence of Tat and its TAR-binding function at an unprecedented resolution. The Tat-TAR interaction is thought to be driven by an enrichment of basic residues in the core of the protein rather than a specific amino acid sequence, but it is not known if point mutations outside of this core region can affect the TAR interaction. This study of mutations that affect the affinity of Tat to TAR can contribute to our understanding of both protein-RNA interactions, as well as the mechanism of HIV transcription and activation.
By creating a library of hundreds of thousands of variants of Tat and selecting for binding to TAR using a yeast three-hybrid assay, we are examining the relationship between the sequence of Tat and its TAR-binding function at an unprecedented resolution. The Tat-TAR interaction is thought to be driven by an enrichment of basic residues in the core of the protein rather than a specific amino acid sequence, but it is not known if point mutations outside of this core region can affect the TAR interaction. This study of mutations that affect the affinity of Tat to TAR can contribute to our understanding of both protein-RNA interactions, as well as the mechanism of HIV transcription and activation.
High throughput proteome-wide search for ubiquitinated proteins in yeast
Lea Starita and Russell Lo (former lab members)
Our goal is to improve upon existing biochemical approaches for finding sites of ubiquitin attachment on all yeast proteins.
Presently, the approach is to purify proteins with an 8xHis-tagged ubquitin followed by tryptic digest to generate peptide fragments for LC/LC-MS/MS analysis. The tryptic fragments that contain a lysine residue that had been ubiquitinated can be identified by a mass shift corresponding to 2 glycine residues left attached to an internal lysine. Historically, only a relatively small number of GG-peptides have been found due to low abundance of GG-peptide as compared to unmodified peptides.
We seek to reduce the complexity of the resulting peptides by using the chemical NTCB (2-nitro-5-thiocyanatobenzoic acid) which cleaves at cysteine residues (Figure 1B). Since ubiquitin does not have any cysteines, the proteome can be cleaved prior to affinity purification ubiquitinated proteins (Figure 1C) and typsinization (Figure 1D). This step will remove many non-ubiquitinated fragments therefore reducing the complexity of the sample. We hope to further decrease the complexity of the peptides by separating forked peptides from linear peptides by strong-cation exchange chromatography (Figure 1E) prior to injection into the mass spectrometer.
Presently, the approach is to purify proteins with an 8xHis-tagged ubquitin followed by tryptic digest to generate peptide fragments for LC/LC-MS/MS analysis. The tryptic fragments that contain a lysine residue that had been ubiquitinated can be identified by a mass shift corresponding to 2 glycine residues left attached to an internal lysine. Historically, only a relatively small number of GG-peptides have been found due to low abundance of GG-peptide as compared to unmodified peptides.
We seek to reduce the complexity of the resulting peptides by using the chemical NTCB (2-nitro-5-thiocyanatobenzoic acid) which cleaves at cysteine residues (Figure 1B). Since ubiquitin does not have any cysteines, the proteome can be cleaved prior to affinity purification ubiquitinated proteins (Figure 1C) and typsinization (Figure 1D). This step will remove many non-ubiquitinated fragments therefore reducing the complexity of the sample. We hope to further decrease the complexity of the peptides by separating forked peptides from linear peptides by strong-cation exchange chromatography (Figure 1E) prior to injection into the mass spectrometer.
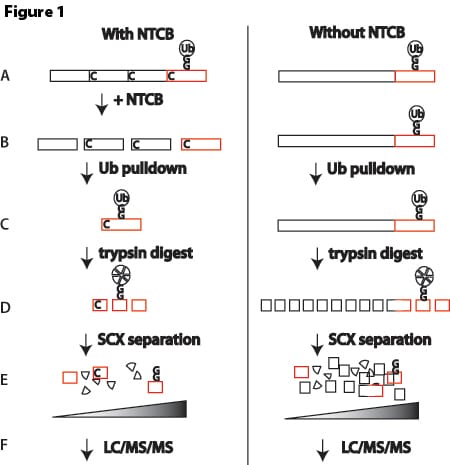
Using the technique outlined above we found a total of 965 unique sites of ubiquitin attachment on 410 yeast proteins. We are able to gain a 1.5X enrichment of GG-peptides using the NTCB cleavage strategy. Most of the improvement in sequencing GG-peptides is achieved by using the high-mass accuracy Orbitrap-LTQ.
Published Results
Starita, L.M., Lo, R.S., Eng, J.K., Von Haller, P.D. and Fields, S. Sites of ubiquitin attachment in Saccharomyces cerevisiae. 2012 Proteomics, Jan;12(2):236-240. Epub 2011 Nov 22
Download PDF
Starita, L.M., Lo, R.S., Eng, J.K., Von Haller, P.D. and Fields, S. Sites of ubiquitin attachment in Saccharomyces cerevisiae. 2012 Proteomics, Jan;12(2):236-240. Epub 2011 Nov 22
Download PDF
Phage Technology
Lysis protein requirements of an RNA phage
Linnea Peterson-Bunker (former lab member)
Lysis of the bacterial host is the critical final step in infection for most phages. The decision of when and how to lyse are key determinants of phage fitness. The best-studied mechanisms of lysis are those of the double-stranded DNA phages, which encode multiple proteins to subvert the different layers of the host cell wall. On the other hand, small, single-stranded RNA phages, such as the leviviruses, encode single lysis proteins as small as 34 amino acids. These phages provide insight into the minimal requirements for phage lysis. Levivirus phage lysis proteins are small, highly diverse, and have evolved multiple times. The only commonality among them is the presence of a transmembrane helix within the protein.
MS2 is a long-studied levivirus with a genome roughly 3.5 kb in size, encoding only 4 genes. Using MS2 as a model, I am determining minimal sequences that function as levivirus lysis proteins, as well as RNA secondary structures used to regulate expression and timing of lysis. Using a cDNA clone of the full-length MS2 genome, I am testing MS2’s tolerance for alternative genome organizations by moving the lysis coding region. These rearrangements destroy the existing secondary structures that regulate translation. By passaging these mutagenized phages, I can see how the phage evolves new regulatory RNA secondary structures or improves upon existing ones.
MS2 is a long-studied levivirus with a genome roughly 3.5 kb in size, encoding only 4 genes. Using MS2 as a model, I am determining minimal sequences that function as levivirus lysis proteins, as well as RNA secondary structures used to regulate expression and timing of lysis. Using a cDNA clone of the full-length MS2 genome, I am testing MS2’s tolerance for alternative genome organizations by moving the lysis coding region. These rearrangements destroy the existing secondary structures that regulate translation. By passaging these mutagenized phages, I can see how the phage evolves new regulatory RNA secondary structures or improves upon existing ones.
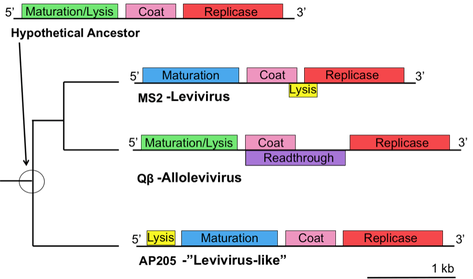
Figure 1. Levivirus lysis proteins have evolved multiple times and exist in a variety of overlapping and non-overlapping genomic locations.

Figure 2. Levivirus lysis proteins vary in size and sequence, with the only commonality being the presence of a transmembrane helix.
Engineering diversity generating retroelements as tools for in vivo targeted mutagenesis
Bryan Andrews
Phages, the most diverse biological entities, are also among the least understood. The vast majority of phage genes have no assigned function, and this uncharted space is likely to contain genes that can inform our understanding of biology or provide unique molecular tools. Diversity Generating Retroelements (DGRs) are small clusters of genes present in many phage and bacterial genomes that diversify a variable region (VR) via reverse transcriptase (RT)-mediated adenine-specific mutagenesis of a template region (TR) transcript, followed by invasion of the mutagenized copy into the homologous VR.
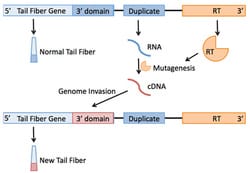
Most work by other labs has focused on the DGR from Bordetella Plus-trophic Phage (BPP-1) in its native context. We would like to engineer the BPP-1 DGR to work in other systems, such as the well-studied phage lambda. By targeting the BPP-1 DGR against the lambda tail fiber, we could create a library of lambda variants, some of which may recognize novel receptors on their host, E. coli. Analyzing these novel receptors will give us insight into how new protein binding interactions evolve, and may provide tricks to improve phage therapy, the treatment of bacterial infections with phages targeting those bacteria.
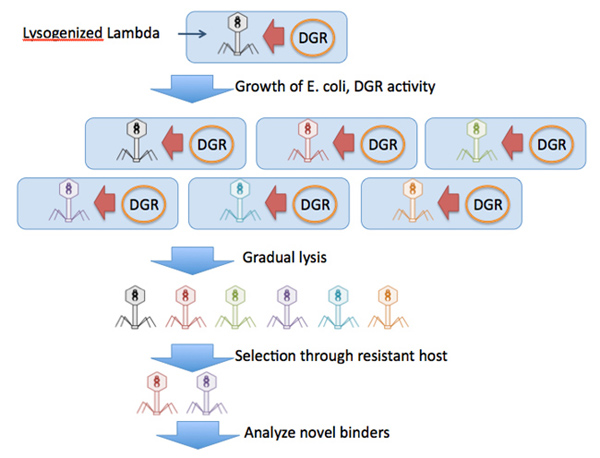
We stopped work on this project to engineer Diversity Generating Retroelements (DGRs) for use in E. coli. Despite several attempts, we were unable to see DGR-specific mutagenesis in vivo. Brt, the relevant reverse transcriptase, was sequestered to inclusion bodies under even modest induction, although it could be solubilized by co-expression of GroES and GroEL. However, this process did not result in functional DGR activity. Pilot attempts at using 6xHis-tagged Brt for in vitro reverse transcription were unsuccessful, but the process did not undergo extensive troubleshooting. For further (mostly negative) data on DGRs, contact bryan92[at]uw.edu.
Testing for diversity generating retroelement activity in yeast
Russell Lo (former lab member)
As researchers, generating protein diversity via saturating mutagenesis of a target gene is a means of exploring protein structure or function through the analysis of the resulting protein variants. In nature, many organisms have evolved robust mechanisms of protein diversification such as the V(D)J recombination system which is required to generate adaptive immunity in vertebrates. Recently, an additional protein diversification mechanism has been discovered in BPP-1 bacteriophage. An encoded retroelement termed DGR (Diversity-Generating Retroelement) can diversify the BPP-1 tail fiber protein that facilitates binding to the bacteriophage’s host, the Bordatella species of bacteria. The retroelement uses an error-prone reverse transcriptase to generated mutations at adenine residues during cDNA synthesis of a template termed TR (Template Region). This mutated copy is then used to swap out a homologous VR (Variable Region) in the coding sequence of the tail fiber gene, Mtd. This retroelement is capable of generating enormous amounts of tail fiber diversity, thus potentially allowing the phage to bind to many different targets on the host cell surface.
The DGR system presents the researcher with an opportunity to introduce huge amounts of diversity into a gene of interest in a novel fashion. To that end, we are attempting to introduce this diversity-generating retroelement into yeast. As a test case for function, we will be tuning the retroelement components for expression in yeast and diversifying the HIS3 gene. cDNA integration will be selected for by replacement of a premature stop codon in HIS3 to restore wildtype function. Adenine mutagenesis will be confirmed via sequencing.
The DGR system presents the researcher with an opportunity to introduce huge amounts of diversity into a gene of interest in a novel fashion. To that end, we are attempting to introduce this diversity-generating retroelement into yeast. As a test case for function, we will be tuning the retroelement components for expression in yeast and diversifying the HIS3 gene. cDNA integration will be selected for by replacement of a premature stop codon in HIS3 to restore wildtype function. Adenine mutagenesis will be confirmed via sequencing.
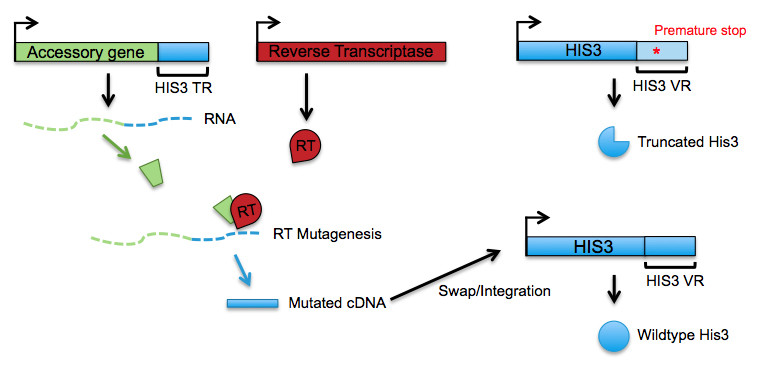
We expressed these DGR components in S. cerevisiae but we never observed adenine mutagenesis.
Synthetic Biology
E. coli biosensors based on ligand-dependent stability
Ben Brandsen
Biosensors are genetically encoded proteins or nucleic acids that enable cells to sense and respond to stimuli. They can be used to control gene expression, to construct genetic circuits, and to detect metabolic products for genome engineering. Many bacterial biosensors are based on natural allosteric transcriptional repressors, which after binding ligand, no longer repress a reporter gene and result in increased gene expression. Engineering allosteric transcription factors to bind new ligands is challenging, however, because engineered transcription factors often have broken allostery.
One general strategy to new biosensors relies upon ligand-dependent protein stabilization. In this biosensor design, a protein that binds to ligand is destabilized with mutations, such that it is rapidly degraded in the absence of ligand, but stabilized in the presence of ligand (Figure 1A). We constructed new E. coli transcriptional biosensors based on ligand-dependent stabilization by fusing the LacI ligand-binding domain between the Zif268 DNA-binding domain and the RpoZ transcription-activating domain. We generated an error-prone PCR library of LacI ligand-binding domains between Zif268 and RpoZ, and subjected this library to a positive selection for growth with ligand (Figure 1B). The biosensor was coupled to the selectable marker HIS3, which was required for growth in the absence of histidine. Variants that survived selection were replica plated on minimal media with and without IPTG ligand, and variants that grew better with ligand compared to without ligand were selected for further analysis.
One general strategy to new biosensors relies upon ligand-dependent protein stabilization. In this biosensor design, a protein that binds to ligand is destabilized with mutations, such that it is rapidly degraded in the absence of ligand, but stabilized in the presence of ligand (Figure 1A). We constructed new E. coli transcriptional biosensors based on ligand-dependent stabilization by fusing the LacI ligand-binding domain between the Zif268 DNA-binding domain and the RpoZ transcription-activating domain. We generated an error-prone PCR library of LacI ligand-binding domains between Zif268 and RpoZ, and subjected this library to a positive selection for growth with ligand (Figure 1B). The biosensor was coupled to the selectable marker HIS3, which was required for growth in the absence of histidine. Variants that survived selection were replica plated on minimal media with and without IPTG ligand, and variants that grew better with ligand compared to without ligand were selected for further analysis.
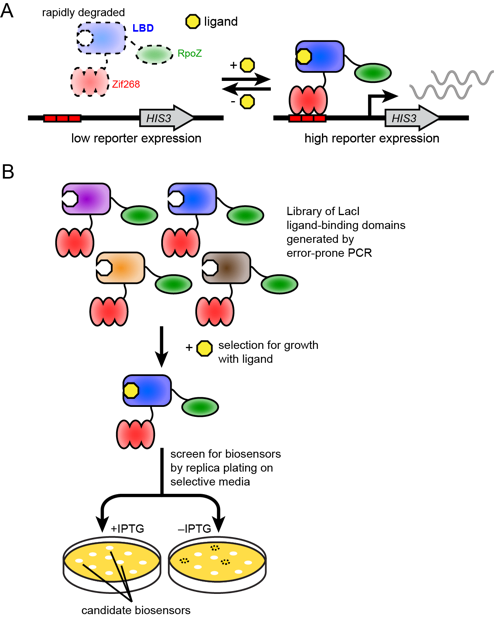
Figure 1. A. Design of biosensors based on ligand-dependent stabilization. The LacI LBD is fused between the Zif268 DBD and RpoZ TAD and destabilized, such that in the absence of ligand, the biosensor is degraded, but in the presence of ligand, the biosensor is stabilized by binding ligand. This biosensor is coupled to a HIS3 reporter gene downstream of a Zif268 binding site. B. Selection strategy to identify new biosensors. An error-prone PCR library of LBD variants is subjected to positive selection with ligand in minimal media lacking histidine, such that only variants able to activate HIS3 expression grow. Biosensors that show ligand-dependent stabilization are subsequently identified by replica plating on minimal media with and without ligand.
We identified a biosensor, L1.0, that shows ligand-dependent expression of HIS3, assessed by increased growth in media lacking histidine (Fig. 2A). It contains six amino acid mutations in the LacI region (Fig. 2B). To determine which amino acid mutations are required for biosensor behavior, we reverted each mutation back to the WT amino acid identity. All amino acid revertants except one, G272R, showed activity similar to L1.0, while reverting G272R abolished biosensor activity (Fig. 2C). We assessed biosensor activity with LacI G272R (named L1.7), and found that it showed a similar ligand-dependent response as L1.0 (Fig. 2D), establishing that a single amino acid mutation is sufficient to confer biosensor behavior. Finally, we confirmed that L1.7 was functioning by ligand-dependent stabilization using Western Blot. We observed an approximately 5-fold increase in the level of L1.7 with 100 mM IPTG compared to without IPTG, verifying increased stability, and therefore increased accumulation, that is dependent on ligand.
Brandsen et al. (2018) ACS Synthetic Biology 7, 1990-1999
Brandsen et al. (2018) ACS Synthetic Biology 7, 1990-1999
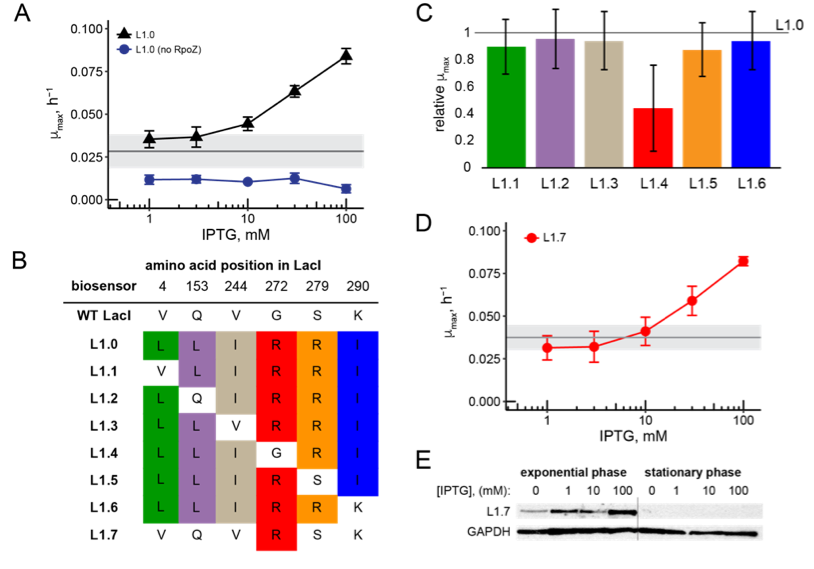
Figure 2. A. Response of L1.0 and L1.0 without RpoZ at increasing IPTG concentration in minimal media. B. Amino acid mutations present in each LacI-based biosensor. C. Response of L1.1-L1.6, relative to L1.0, when induced with 100 mM IPTG. D. Response of biosensor L1.7 at increasing IPTG concentration in minimal media. E. Ligand-dependent accumulation of L1.7 as assessed by Western blot.
Engineering Natural Product Biosynthetic Pathways with Multiplexed Mutagenesis and Biosensor-Based Selection
Ben Brandsen (former lab member)
Antibiotic resistance is a serious and growing problem. In 2013, the U.S. Centers for Disease Control reported that antibiotic resistance is responsible for more than 23,000 deaths and 2 million infections each year, costing approximately $20 billion. The problem of increasing antibiotic resistance is compounded by the small number of new antibiotics approved by the FDA in the past decade and a dearth of new antibiotic drug candidates in clinical trials. One rich source of antibiotic lead compounds is natural products, but isolation of new natural products has slowed, significantly hampered by the rediscovery of known compounds. If the biosynthetic pathways that synthesize therapeutically useful natural products can be efficiently engineered in a laboratory, then new, natural product-like compounds would be accessible.
Polyketides are a class of natural products from which many important antibiotic compounds have been identified, including erythromycin, azithromycin, and clarithromycin. Polyketides are biosynthesized by polyketide synthase (PKS) enzymes, which consist of several modular protein domains that iteratively add small extender substrates together. One of the most well-studied polyketide biosynthetic pathways produces the antibiotic erythromycin A (Figure 1). It consists of three synthases, named DEBS1-3. The synthases contains two modules, each of which incorporates a single extender substrate. A thioesterase domain on DEBS3 cyclizes the polyketide to form 6-deoxyerythronolide B, and additional tailoring enzymes convert 6-deoxyerythronolide B (6-dEB) to erythromycin A.
Polyketides are a class of natural products from which many important antibiotic compounds have been identified, including erythromycin, azithromycin, and clarithromycin. Polyketides are biosynthesized by polyketide synthase (PKS) enzymes, which consist of several modular protein domains that iteratively add small extender substrates together. One of the most well-studied polyketide biosynthetic pathways produces the antibiotic erythromycin A (Figure 1). It consists of three synthases, named DEBS1-3. The synthases contains two modules, each of which incorporates a single extender substrate. A thioesterase domain on DEBS3 cyclizes the polyketide to form 6-deoxyerythronolide B, and additional tailoring enzymes convert 6-deoxyerythronolide B (6-dEB) to erythromycin A.
Figure 1. Biosynthesis of erythromycin by DEBS1-3. 6-dEB is modified by additional tailoring enzymes to generate the natural product antibiotic erythromycin A.
Because of the iterative manner by which polyketides are built and the modular nature of the PKS enzymes, engineering the biosynthetic pathways to produce new compounds is an appealing prospect. Efforts have primarily focused on changing the order of protein domains, substituting exogenous enzyme domains, and supplying unique extender substrates. However, technical challenges, such as poor interaction between non-native protein domains and reduced enzymatic activity, have stalled these efforts.
I hope to overcome many of these challenges by using high-throughput DNA mutagenesis and biosensor-based selection to improve the activity of chimeric PKS enzymes. I have established that allosteric transcription factor MphR, which activates expression of a downstream gene in the presence of erythromycin, can be used for ligand-dependent expression of a HIS3 reporter gene. I plan to build a library of chimeric DEBS pathways with acyltransferase domains from other polyketide pathways (as shown in Figure 2), which control the substrates that are incorporated into the growing polyketide, and connect production of their macrolide ligand to the response of MphR.
I hope to overcome many of these challenges by using high-throughput DNA mutagenesis and biosensor-based selection to improve the activity of chimeric PKS enzymes. I have established that allosteric transcription factor MphR, which activates expression of a downstream gene in the presence of erythromycin, can be used for ligand-dependent expression of a HIS3 reporter gene. I plan to build a library of chimeric DEBS pathways with acyltransferase domains from other polyketide pathways (as shown in Figure 2), which control the substrates that are incorporated into the growing polyketide, and connect production of their macrolide ligand to the response of MphR.
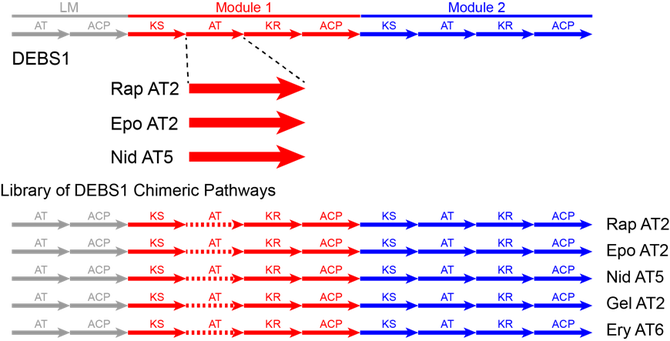
Figure 2. Strategy for constructing DEBS chimeras. Acyltransferase (AT) domains from several other polyketide biosynthetic pathways that incorporate different substrates into the polyketide natural product are swapped for the native AT domain, to generate a library of chimeric synthases.
Finally, I will use multiplexed genome engineering in E. coli to introduce mutations near the substitute AT domain, and select for active pathway variants using the MphR biosensor to detect product formation (Figure 3). This strategy of high-throughput mutagenesis and biosensor-based selection could also be applied to other biosynthetic pathways that produce valuable small molecule or protein products.
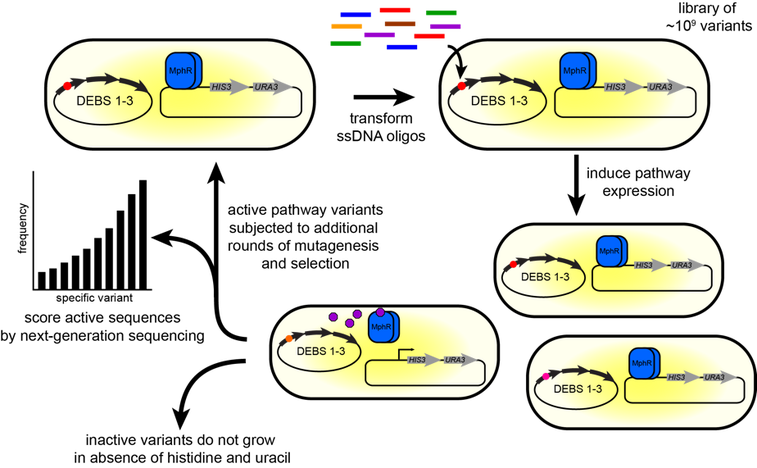
Figure 3. Biosensor-based strategy to improve biosynthetic pathway production. DEBS production is coupled to the MphR transcription factor, which in the presence of ligand, increases expression of the HIS3 and URA3 reporter genes required for growth in minimal media lacking histidine and uracil. After in vivo mutagenesis using DNA oligos, pathway variants able to produce the macrolide product will grow faster than those that produce no product or less product, such that after selection, active pathway variants make up a large fraction of the cell population. These will be scored by deep sequencing, to identify the best mutations, and subjected to additional rounds of mutagenesis and selection.
Using yeast counter-selection for the improved development of ligand-stabilized biosensor
Matt Rich (former lab member)
Yeast and other microbes can be used to produce many small molecules, but optimization of the metabolic pathways involved is a non-trivial undertaking. Strains optimized to produce a small molecule of interest can be selected through the use of suitable biosensors, but few of these are currently available. As such, we must design new sensors for use in strain engineering contexts. We have previously shown that ligand-stabilized transcription factors are a viable, and likely general, biosensor platform. In this approach (Figure 1), an unstable transcription factor can induce expression of a reporter gene only in the presence of a stabilizing ligand. Optimization of this stabilizing interaction can be difficult, and would be aided by an in vivo selection and counter-selection strategy that enables the use of large populations of protein variants.
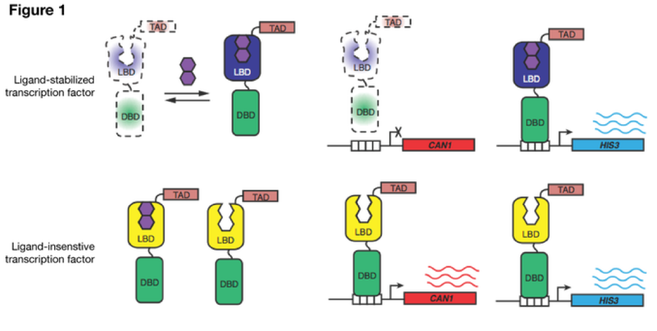
We have implemented such a strategy using the yeast CAN1 gene. CAN1 encodes an arginine transporter that also imports canavanine, a toxic arginine analog, into the cell. Because evidence has shown that low expression of CAN1 is not fully toxic in the presence of canavanine, we hypothesized that this gene could be used as a counter-selectable marker for ligand-stabilized transcription factor optimization. Indeed, increased biosensor stabilization causes a growth defect in the presence of canavanine (Figure 2). We are currently testing selection methods in which we initially positively select for HIS3 expression in the presence of ligand, followed by counter-selection in canavanine in the absence of ligand.
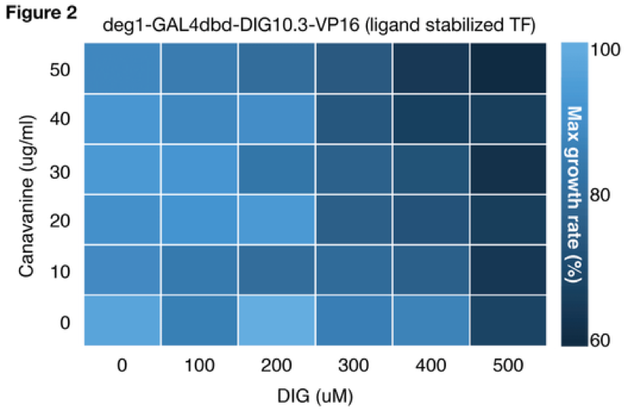
The use of CAN1 has drawbacks, though. For instance, the mutation rate at the CAN1 locus is relatively high, and loss-of-function mutations in CAN1 break the counter-selection. We are exploring the use of diploid reporter strains, which should diminish this problem. CAN1 seems to require a higher expression level for phenotypic effects when compared to the positive selectable marker, HIS3.
Genetic sensors for the detection of biosynthetic product
Ben Jester (former lab member
Model microorganisms such as E. coli and S. cerevisiae are valuable tools for the production of small molecules, such as therapeutics and biofuels, but the optimization of biosynthetic pathways in a new host is non-trivial. Identifying genetic modifications that enhance metabolite synthesis can be an exceptionally laborious process, particularly in the absence of a method to easily determine product yields. Genetically encoded biosensors that couple small molecule recognition to a readily measured output allow more rapid identification of cells with enhanced biosynthetic production or conditions that promote enhanced production. One of Nature’s most common mechanisms for detecting the presence of a small molecule – be it a metabolite or an environmental agent – has been to evolve biosensors that regulate the transcription of one or more genes upon binding of the relevant ligand. The goal of this project is to design a transcription factor (TF)-based strategy for biosensors that may provide a general solution to the problem of small molecule detection.
Our strategy is to use destabilizing mutations that impair functional expression of the biosensor until it binds its cognate ligand. It should be possible to design a protein-based biosensor for any ligand as long as a ligand-binding domain (LBD) exists – or can be designed – for the desired small molecule. By fusing an unstable LBD to a TF, we couple ligand-dependent stabilization to reporter gene expression. As a proof of principle, we chose to use a de novo designed LBD, DIG10.3, which binds digoxigenin, a steroid similar to drugs used to treat heart failure. By fusing this protein to a DNA-binding domain (DBD) and a transcriptional activation domain (TAD), we were able to generate a ligand-dependent TF, which we designated GDVP. From a library of mutants, FACS analysis allowed us to identify destabilizing mutations that improved sensor function by >10-fold (GDVP.1 and GDVP.2).
Our strategy is to use destabilizing mutations that impair functional expression of the biosensor until it binds its cognate ligand. It should be possible to design a protein-based biosensor for any ligand as long as a ligand-binding domain (LBD) exists – or can be designed – for the desired small molecule. By fusing an unstable LBD to a TF, we couple ligand-dependent stabilization to reporter gene expression. As a proof of principle, we chose to use a de novo designed LBD, DIG10.3, which binds digoxigenin, a steroid similar to drugs used to treat heart failure. By fusing this protein to a DNA-binding domain (DBD) and a transcriptional activation domain (TAD), we were able to generate a ligand-dependent TF, which we designated GDVP. From a library of mutants, FACS analysis allowed us to identify destabilizing mutations that improved sensor function by >10-fold (GDVP.1 and GDVP.2).
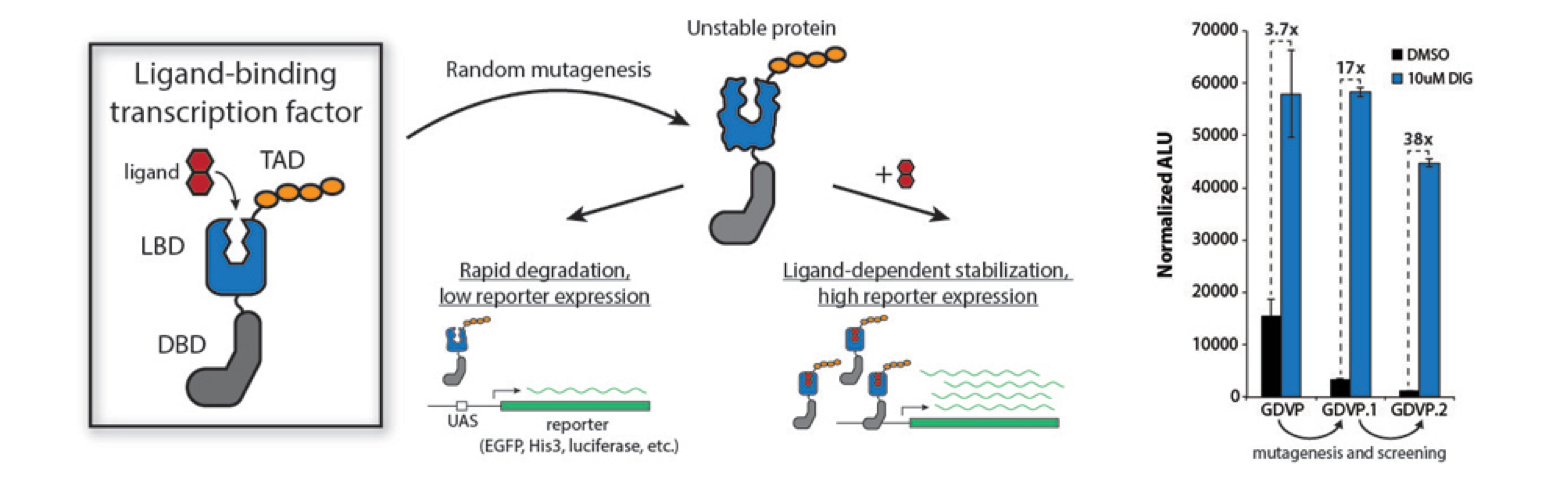
In order to tune the sensor for selections using a HIS3 reporter, we fused the degron from the Mata protein to GDVP to increase degradation. The fusion generated a sensor that leads to growth in histidine-deficient media more than 3 orders of magnitude better when the ligand (digoxigenin) is present. Sensitivity to digoxigenin can be further improved by deleting yeast efflux pumps, like Pdr5.
While the biosynthetic pathway required to produce digoxigenin is not known, other steroids such as progesterone have been successfully produced by yeast. An additional round of mutagenesis and screening allowed us to create a sensor that is extremely selective for progesterone but not its biosynthetic precursor, pregnenolone. Collaborators have used this sensor to identify mutations in 3-beta-hydroxysteroid dehydrogenase that improve progesterone bioproduction in yeast. We are currently applying these methods to develop sensors for new small molecules
While the biosynthetic pathway required to produce digoxigenin is not known, other steroids such as progesterone have been successfully produced by yeast. An additional round of mutagenesis and screening allowed us to create a sensor that is extremely selective for progesterone but not its biosynthetic precursor, pregnenolone. Collaborators have used this sensor to identify mutations in 3-beta-hydroxysteroid dehydrogenase that improve progesterone bioproduction in yeast. We are currently applying these methods to develop sensors for new small molecules
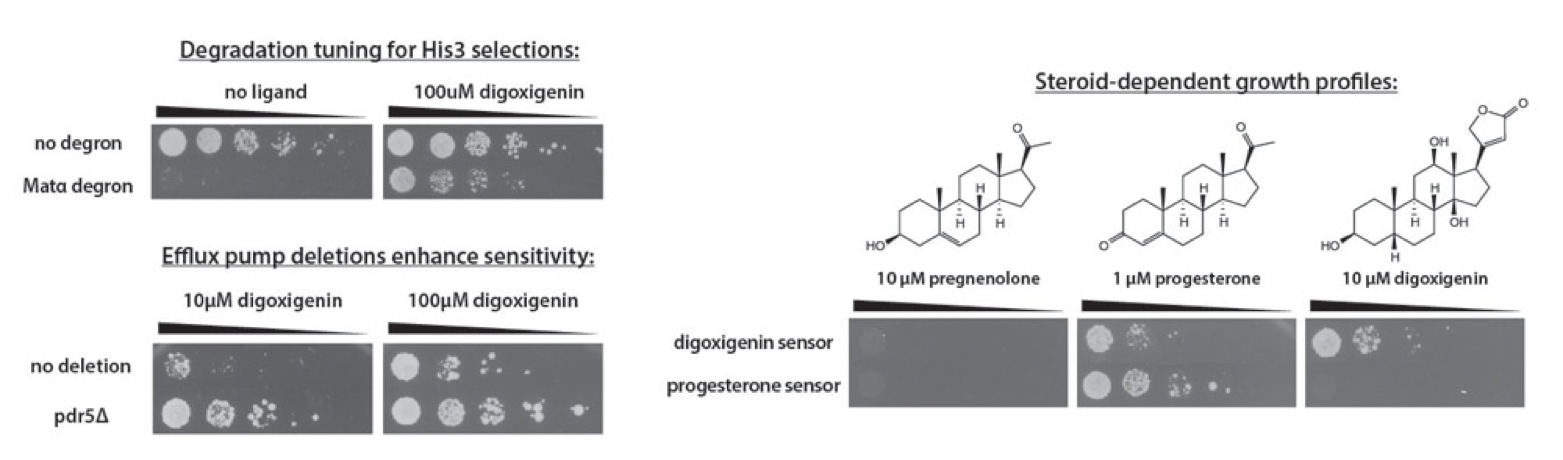
Engineering ligand-gated dimers to control cellular function
Ben Jester (former lab member
Protein-protein interactions drive the overwhelming majority of cellular processes, such as signaling, differentiation, and proliferation. Interacting protein domains coordinate these critical functions by ensuring that functional domains act on the appropriate target. Native interacting domains can often be replaced with an alternative pair of interacting proteins that result in near wild-type function. For example, the protein pair of FKBP and FRB is a popular substitute for interacting domains because interaction between these two proteins is dependent upon the small molecule rapamycin. When fused to functional domains requiring co-localization for activity, they provide the researcher with temporal control over a cellular process. Currently, the toolkit for small molecule-gated protein-protein interactions is exceptionally small. As the field of synthetic biology continues to grow, so will the need for tools that provide temporal control over multiple cellular processes.
To approach this problem, we have taken an unstable, homodimeric ligand-binding domain (LBD) and converted it into a ligand-dependent dimer. The LBD used here, DIG10.3, is a small protein that is degraded by the cell until it binds to the small molecule digoxigenin. To measure LBD dimerization, we made two fusion proteins: a single monomer of the LBD fused to a DNA-binding domain (DBD), and a single monomer of the LBD fused to a transcriptional activation domain (TAD). In an assay conceptually similar to the yeast two-hybrid system, dimerization between these two fusion proteins should induce the expression of a reporter protein. Previous work with this protein has shown that mutations to the dimer interface of DIG10.3 can lead to destabilization of the protein. By testing pairs of proteins that contained mutations at the dimer interface or in the ligand-binding pocket, we were able to identify dimers that respond to either digoxigenin or progesterone, or that required both ligands to activate reporter expression.
To approach this problem, we have taken an unstable, homodimeric ligand-binding domain (LBD) and converted it into a ligand-dependent dimer. The LBD used here, DIG10.3, is a small protein that is degraded by the cell until it binds to the small molecule digoxigenin. To measure LBD dimerization, we made two fusion proteins: a single monomer of the LBD fused to a DNA-binding domain (DBD), and a single monomer of the LBD fused to a transcriptional activation domain (TAD). In an assay conceptually similar to the yeast two-hybrid system, dimerization between these two fusion proteins should induce the expression of a reporter protein. Previous work with this protein has shown that mutations to the dimer interface of DIG10.3 can lead to destabilization of the protein. By testing pairs of proteins that contained mutations at the dimer interface or in the ligand-binding pocket, we were able to identify dimers that respond to either digoxigenin or progesterone, or that required both ligands to activate reporter expression.
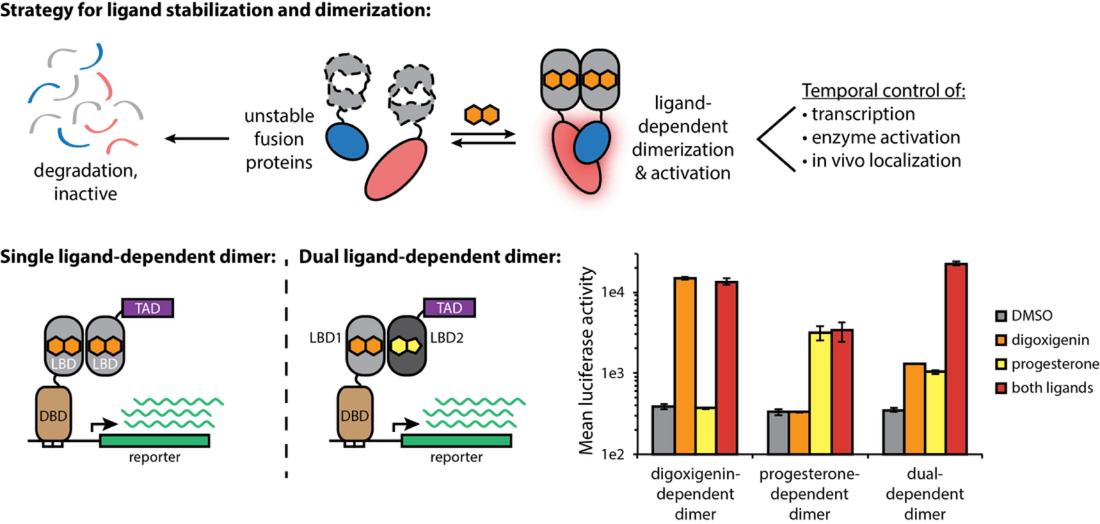
The use of dimeric proteins fused to two different components (i.e. a DNA-binding domain and a transcriptional activation domain) is limited by the possibility of non-functional dimers that can form between identical components. Using a crystal structure model of the DIG10.3 homodimer, we made several rational mutations to the dimer interface that were predicted to favor heterodimerization of the two proteins. Two redesigns based on either a “charge-swap” (1a and 1b) or “knob-in-hole” (2a and 2b) model generated a pair of orthogonal interfaces and improved inducible reporter activation by more than 10-fold. On-going work is focused on exploring how directed evolution and deep sequencing may allow us to identify additional unique interfaces and deploying multiple pairs of ligand-dependent dimers in the same cell.
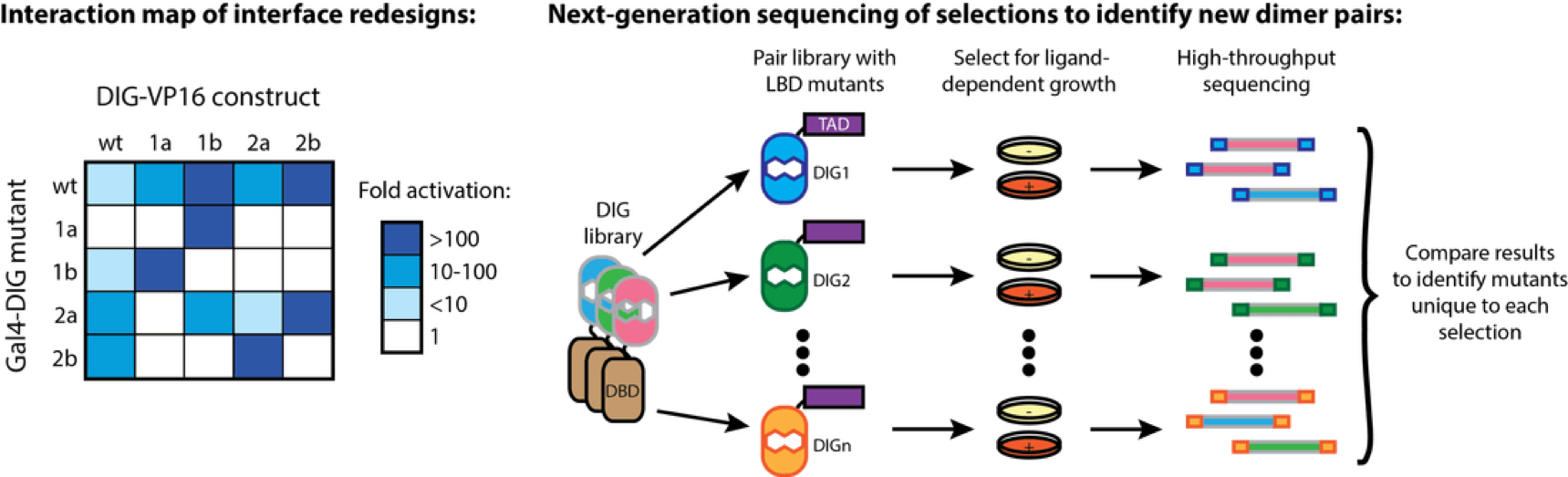
Engineering a biosensor for 3’-5’ Phosphoadenosine phosphate
Wei Zhou
3’-5’ Phosphoadenosine phosphate (pAp) is produced by removal of a sulfate group from 3’-phosphoadenosine 5’-phosphosulfate (PAPS), which is the most common coenzyme in the sulfotransferase reaction. pAp is converted into AMP and Pi by a pAp phosphatase (Met22). The pAp level in the cell is normally not detectable; however, pAp accumulates when Met22 activity is impaired. The accumulation of pAp is toxic to the tRNA quality control pathway by inhibiting a critical RNA processing enzyme activity.
A biosensor for pAp concentration would both indicate whether the tRNA quality control machinery is active, and report on the methionine synthesis pathway. I am using protein engineering to create such a biosensor by generating a hybrid protein that contains a DNA-binding domain (DBD), a transcriptional activation domain (TAD) and a pAp ligand-binding domain (LBD). I will destabilize this protein via mutation, such that it will be degraded and not activate transcription. Binding of pAp will stabilize the hybrid protein and turn on expression of the reporter gene, GFP, thus reporting on the level of pAp.
I have selected part of the Met22 protein as a ligand-binding domain for pAp and confirmed that this domain has lost catalytic activity. I have mutagenized this domain to generate a library that is fused to a transcription activation domain and a DNA-binding domain. The library is transformed into a yeast strain with a GFP reporter gene that binds to the DBD. By sorting for fluorescence under conditions of either high pAp or low pAp, I should be able to identify an appropriate biosensor, and characterize the mutations present in it.
A biosensor for pAp concentration would both indicate whether the tRNA quality control machinery is active, and report on the methionine synthesis pathway. I am using protein engineering to create such a biosensor by generating a hybrid protein that contains a DNA-binding domain (DBD), a transcriptional activation domain (TAD) and a pAp ligand-binding domain (LBD). I will destabilize this protein via mutation, such that it will be degraded and not activate transcription. Binding of pAp will stabilize the hybrid protein and turn on expression of the reporter gene, GFP, thus reporting on the level of pAp.
I have selected part of the Met22 protein as a ligand-binding domain for pAp and confirmed that this domain has lost catalytic activity. I have mutagenized this domain to generate a library that is fused to a transcription activation domain and a DNA-binding domain. The library is transformed into a yeast strain with a GFP reporter gene that binds to the DBD. By sorting for fluorescence under conditions of either high pAp or low pAp, I should be able to identify an appropriate biosensor, and characterize the mutations present in it.
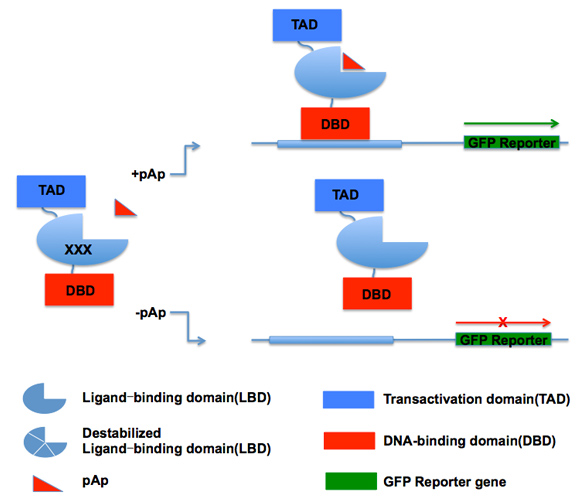
Engineer a biosensor for detection of HPV16-L1 production
Wei Zhou
HPV human papillomaviruses (HPV) are common pathogens associated with a variety type of cancers, especially cervical cancer, which is the second most common cancer in women in United States. HPV16 infection is responsible for up to 50% of all cervical cancers.
Successful preventive measures for HPV16 infection rely on vaccination. The vaccine is a virus-like particles generated from HPV16-L1 protein. When manufacturing the vaccine, viral capsid L1 gene is incorporated in yeast genome first and then the yeast produces HPV16 L1 capsid protein and it will be self-assembled to virus-like particles (vaccine). For many years, people are interested in using metabolic engineering the yeast genome to increase the production of L1 capsid protein. My project focus on engineering a biosensor for L1 capsid protein and using high-throughput way to screen those genome mutants which could increase L1 production.
My design strategy is to use destabilizing mutations that impair functional expression of the biosensor until it binds to L1. To do that, I tried HPV-16 L2 protein as the ligand-binding domain for L1. By fusing it with an activation domain and DNA-binding domain, we couple ligand-dependent stabilization to reporter gene expression.
Successful preventive measures for HPV16 infection rely on vaccination. The vaccine is a virus-like particles generated from HPV16-L1 protein. When manufacturing the vaccine, viral capsid L1 gene is incorporated in yeast genome first and then the yeast produces HPV16 L1 capsid protein and it will be self-assembled to virus-like particles (vaccine). For many years, people are interested in using metabolic engineering the yeast genome to increase the production of L1 capsid protein. My project focus on engineering a biosensor for L1 capsid protein and using high-throughput way to screen those genome mutants which could increase L1 production.
My design strategy is to use destabilizing mutations that impair functional expression of the biosensor until it binds to L1. To do that, I tried HPV-16 L2 protein as the ligand-binding domain for L1. By fusing it with an activation domain and DNA-binding domain, we couple ligand-dependent stabilization to reporter gene expression.
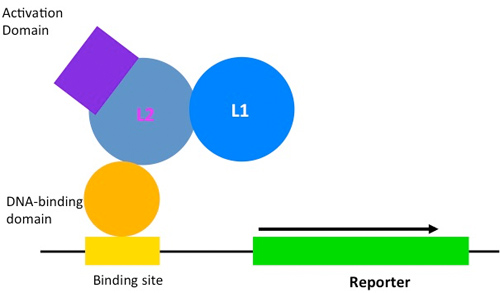
Multiplexed genetic engineering in Saccharomyces cerevisiae
Josh Cuperus and Russell Lo (former lab member)
Strategies to optimize a metabolic pathway often require a large collection of strains to be generated, each containing different versions of the sequences that regulate the expression of the relevant pathway genes. Here we develop a set of reagents and methods to carry out this process at high efficiency in the yeast Saccharomyces cerevisiae. This toolkit includes a set of variants of the tet operator, which in conjunction with a TetR-VP16 activator drive expression over a 100-fold range; the induction of the I-OnuI homing endonuclease to target its recognition site in a gene to be modified, which boosts homologous recombination more than 10^5 over that in the absence of a double-strand break; and the generation of a plasmid carrying the six variant tet operator sites flanked by I-OnuI sites, uncoupling the transformation and recombination steps. As proof of principle, we introduce into the S. cerevisiae genome the three crt genes from Xanthophyllomyces dendrorhous required for yeast to synthesize lycopene, and carry out the recombination process to produce a population of cells with permutations of tetO variants regulating the three genes. We identify 0.7% of the cells as making lycopene, of which the vast majority have undergone a recombination event at each of the crt genes. Based on sequence analysis of these genes in strains that do not produce lycopene, we estimate a rate of ~20% recombination per targeted site, much higher than obtained in other studies.
Published Results
Cuperus JT, Lo RS, Shumaker L, Proctor J, Fields S. A tetO Toolkit To Alter Expression of Genes in Saccharomyces cerevisiae. ACS Synth Biol. 2015 Mar 17. [Epub ahead of print]
Download PDF
Cuperus JT, Lo RS, Shumaker L, Proctor J, Fields S. A tetO Toolkit To Alter Expression of Genes in Saccharomyces cerevisiae. ACS Synth Biol. 2015 Mar 17. [Epub ahead of print]
Download PDF
Metabolites
An integrated metabolomic approach to understanding drug function
Doug Fowler and Jason Stephany (former lab members)
Metabolites are a unique and highly diverse class of elements and compounds that constitute the “business end” of biochemistry. For example, the budding yeast S. cerevisiae is estimated to contain thousands of unique metabolites at a wide range of concentrations. In addition to being both substrates for and products of protein action, metabolites have profound regulatory effects ranging from simple enzymatic product inhibition to allostery to initiation of complex signaling cascades that regulate gene expression programs. Furthermore, exogenous metabolites, acquired for nutritive purposes or used as chemical defenses, greatly expand the diversity of metabolites a cell might encounter. Thus, we hypothesized that examination of the effect of excess metabolites on a drug phenotype could provide rich, systems-level information about both cellular and drug function.
To test this principle, we screened a small pilot library of about 50 metabolites in a yeast-based model against lovastatin. Statin drugs inhibit HMG-CoA reductase, which is the rate-limiting enzyme in the synthesis of cholesterol. Consequently, they are among the most widely prescribed drugs in the world, used to treat high cholesterol and atherosclerosis. We chose to investigate statins because, despite being one of the first drugs designed with a specific molecular target in mind, statins have poorly understood pleiotropic effects. For example, in addition to lowering cholesterol by inhibiting HMG-CoA reductase, statins can reduce the risk of death from stroke. Statins can also have significant side effects including musculoskeletal deterioration and rhabdomyolysis, but how these deleterious effects occur is not known.
Statins are effective in inhibiting the yeast orthologs of HMG-CoA reductase and lowering levels of the yeast cholesterol analog, ergosterol. Statin treatment produces dose-dependent growth inhibition in yeast, presumably owing to the requirement for ergosterol for generation of new membrane. We screened our pilot metabolite library against a S. cerevisiae model of statin action. Our metabolite-statin screen revealed that the divalent metal ions zinc, copper and manganese were all effective in alleviating statin mediated growth inhibition. We characterized metal mediated statin rescue using an integrated approach that included biochemical, metabolomic and genomic approaches.
To test this principle, we screened a small pilot library of about 50 metabolites in a yeast-based model against lovastatin. Statin drugs inhibit HMG-CoA reductase, which is the rate-limiting enzyme in the synthesis of cholesterol. Consequently, they are among the most widely prescribed drugs in the world, used to treat high cholesterol and atherosclerosis. We chose to investigate statins because, despite being one of the first drugs designed with a specific molecular target in mind, statins have poorly understood pleiotropic effects. For example, in addition to lowering cholesterol by inhibiting HMG-CoA reductase, statins can reduce the risk of death from stroke. Statins can also have significant side effects including musculoskeletal deterioration and rhabdomyolysis, but how these deleterious effects occur is not known.
Statins are effective in inhibiting the yeast orthologs of HMG-CoA reductase and lowering levels of the yeast cholesterol analog, ergosterol. Statin treatment produces dose-dependent growth inhibition in yeast, presumably owing to the requirement for ergosterol for generation of new membrane. We screened our pilot metabolite library against a S. cerevisiae model of statin action. Our metabolite-statin screen revealed that the divalent metal ions zinc, copper and manganese were all effective in alleviating statin mediated growth inhibition. We characterized metal mediated statin rescue using an integrated approach that included biochemical, metabolomic and genomic approaches.
Published Results
Fowler DM, Cooper SJ, Stephany JJ, Hendon N, Nelson S and Fields S. Suppression of statin effectiveness by copper and zinc in yeast and human cells. Mol. BioSyst. 2011, Feb;7(2):533-544.
Download PDF
Fowler DM, Cooper SJ, Stephany JJ, Hendon N, Nelson S and Fields S. Suppression of statin effectiveness by copper and zinc in yeast and human cells. Mol. BioSyst. 2011, Feb;7(2):533-544.
Download PDF
Metabolite profiling in yeast
Sara Cooper and Sven Nelson (former lab members)
Metabolism encompasses all the processes by which a cell generates energy and other essential molecules from nutrients. These pathways rely on hundreds of genes and involve thousands of small molecule intermediates, vitamins and cofactors. Interest in these molecules has led to development of technologies that allow high-throughput profiling of metabolic intermediates. We have optimized capillary electrophoresis methods for profiling amines, thiols and organic acids in the yeast Saccharomyces cerevisiae. Using these protocols, we have screened the yeast deletion collection and shown that clustering based on metabolite profiles allows us to identify related genes and pathways.
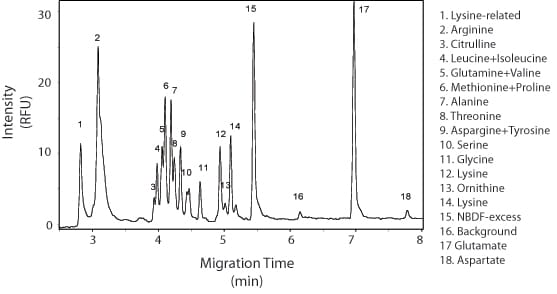
Figure 1. Amino acid profiling of a wild-type yeast extract using fluorescent derivatization of amine groups in combination with capillary electrophoresis separation.
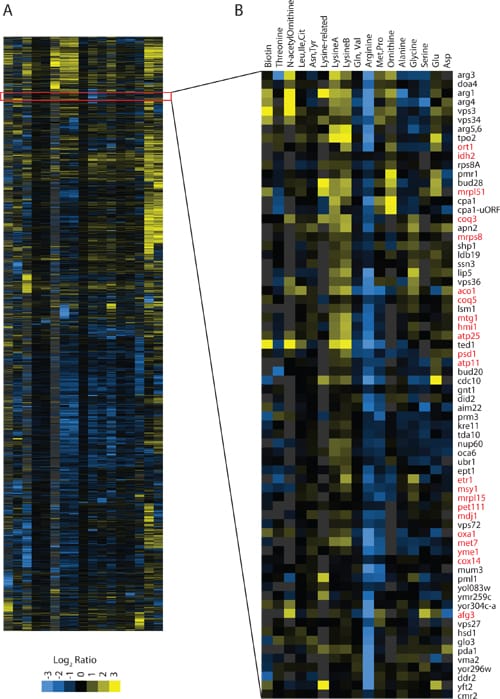
Figure 2. Panel A shows amino acid profiling of the yeast deletion collection clustered by common metabolite profile. Panel B shows that arginine mutants show low levels of arginine and accumulation of arginine precursors such as ornithine and lysine. This cluster is also enriched for mitochondrial genes. Since arginine biosynthesis occurs in the mitochondria we propose that genes affecting mitochondrial function also affect arginine biosynthesis.
We have also begun metabolite profiling using gas chromatography and mass spectrometry (GCxGC-TOF). Preliminary experiments demonstrate that we can identify hundreds of unique compounds, including amino acids, sugars, organic acids and sterols. We are currently applying this method to better understand sterol biosynthesis in yeast. These complementary approaches provide a systematic view of metabolism in yeast. Other applications of this technology that we are working on include metabolite profiling in human urine samples from individuals with kidney disease and characterizing natural variation in yeast by assaying metabolic profiles along with transcription and protein levels in wild yeast strains.
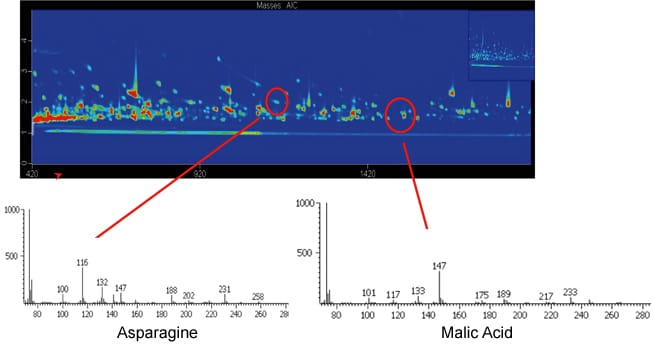
Figure 3. Two-dimensional gas chromatography with mass spectrometry is used for identification and quantification of a couple of hundred intracellular small molecule metabolites.
Published Results
Cooper SJ, Finney GL, Brown SL, Nelson SK, Hesselberth J, Maccoss MJ, Fields S. High-throughput profiling of amino acids in strains of the Saccharomyces cerevisiae deletion collection. Genome Res. 2010 Sep;20(9):1288-96.
Download PDF
Cooper SJ, Finney GL, Brown SL, Nelson SK, Hesselberth J, Maccoss MJ, Fields S. High-throughput profiling of amino acids in strains of the Saccharomyces cerevisiae deletion collection. Genome Res. 2010 Sep;20(9):1288-96.
Download PDF
Projects Completed Before 2010
Genomewide identification of transcription factor binding sites by DNAseI footprinting
Jay Hesselberth and Zhihong Zhang (former lab members)
The complement of DNA-binding proteins and their occupancy of sites throughout the genome determine an organism’s programs of gene expression, DNA replication and other chromosome-based processes. A detailed picture of factor binding on a genome-wide basis exists for Saccharomyces cerevisiae, obtained by a combination of transcriptional profiles, chromatin immunoprecipitation of more than 200 transcription factors, computational analyses and other assays. In an alternative approach, we have used digestion of chromatin by DNase I followed by high throughput DNA sequencing to identify sites of increased nuclease accessibility throughout the yeast genome. The resulting set of more than 10 million sequence reads provides both a global view of chromatin architecture as well as a gene-by-gene view of regulatory sequences protected from digestion by the presence of bound proteins. Unlike the case with results from chromatin immunoprecipitation, these gene-by-gene DNase I footprints can be used to directly identify transcription factor binding sites, and thereby infer their motifs. We found previously unknown binding sites in the genome for well-characterized factors, and observed other annotated binding sites that appear not to be protected from nuclease digestion under our conditions. This approach has the potential to characterize the transcriptional regulatory network of a poorly characterized organism given only its genome sequence.
Published Results
Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, Fields S, Stamatoyannopoulos JA. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009 Apr;6(4):283-9.
Download PDF
Published Results
Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, Fields S, Stamatoyannopoulos JA. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009 Apr;6(4):283-9.
Download PDF
DrnI is a novel debranching enzyme-associated nuclease with a role in intron turnover
Jay Hesselberth (former lab member)
The turnover of introns spliced from pre-mRNA occurs first by debranching the lariat intron followed by destruction of the linear intron by other nucleases. We have identified a novel component of intron turnover, DRN1 (YGR093W). Using yeast two-hybrid screens, we found that Drn1 interacts with the debranching enzyme, Dbr1, and another spliceosomal component, Syf1. Sequence alignments revealed that Drn1 is homologous to the metallophosterase domain of Dbr1, and Drn1 has RNA endonuclease activity in vitro. Deletion of DRN1 results in the accumulation of lariat introns spliced from some ribosomal protein genes. We have identified genetic interactions between DRN1 and mutant alleles of PRP43, suggesting that Drn1 plays a role in the turnover of the spliceosomal complexes containing lariat introns. Intriguingly, the subset of Drn1-effected introns use RNA structural elements to stabilize conformations productive for splicing. We propose a model in which the nuclease activity of Drn1 is required for the efficient turnover of these large, structured introns, whose hyperstability may hinder the dissociation of lariat intron complexes by the spliceosomal DEAD/H-box ATPases.
Yeast protein interaction
Sara Cooper and Sven Nelson (former lab members)
We constructed an array of ~6000 yeast transformants, each designed to express one of the S. cerevisiae open reading frames as a fusion to the Gal4 activation domain (AD). Using robotics, we can carry out a genomewide two-hybrid screen. A yeast strain expressing Gal4 DNA-binding domain (DBD) fused to any protein of interest is mated to the transformants in the array. Diploid cells are selected, pinned onto media selective for the two-hybrid interaction (-histidine) and scored for growth . Only cells that express interacting proteins should grow in the selective plates. The identity of interacting proteins is revealed by the positions of His+ colonies in the array. The initial description of the array, along with a collaborative effort by CuraGen, Inc. to carry out high throughput two-hybrid screens of yeast proteins, is described in Uetz et al. (2000).
The data set can be downloaded here.
To date we have screened over 1000 yeast and several non-yeast proteins against the array. We are continuing this work on a collaborative basis through the Yeast Resource Center (YRC). Researchers with a specific gene of interest can contact us via the YRC website to request a collaboration. The gene of interest needs to be cloned into an appropriate two-hybrid DBD vector that we can provide. The identification of putative protein-protein interactions should further guide investigation of function.
In addition, we are working towards the development of a ‘dual-reporter’ Y2H system that should allow us to eliminate technical false positives that do not arise from a true interaction between the bait and prey proteins. Unlike the current 2-hybrid reporter system, this second reporter is not based on transcriptional activation, but rather on the reconstitution of a protein whose function can be selected or screened for. N-terminal and C-terminal fragments of proteins such as GFP and luciferase have been previously shown to function in split-reporter systems. We are in the process of fusing these domains to a well-established yeast two-hybrid protein pair and are currently testing different combinations in order to identify the optimal conditions that will allow us to distinguish between true and false positives.
Other publications describing yeast protein interactions that include work from the laboratory are listed here.
Select the links below to learn about how the two-hybrid systems works:
How to clone 6000 ORFs
Primers
Vectors: pOBD2 and pOAD
Yeast strains
Recombination Cloning Protocols
The data set can be downloaded here.
To date we have screened over 1000 yeast and several non-yeast proteins against the array. We are continuing this work on a collaborative basis through the Yeast Resource Center (YRC). Researchers with a specific gene of interest can contact us via the YRC website to request a collaboration. The gene of interest needs to be cloned into an appropriate two-hybrid DBD vector that we can provide. The identification of putative protein-protein interactions should further guide investigation of function.
In addition, we are working towards the development of a ‘dual-reporter’ Y2H system that should allow us to eliminate technical false positives that do not arise from a true interaction between the bait and prey proteins. Unlike the current 2-hybrid reporter system, this second reporter is not based on transcriptional activation, but rather on the reconstitution of a protein whose function can be selected or screened for. N-terminal and C-terminal fragments of proteins such as GFP and luciferase have been previously shown to function in split-reporter systems. We are in the process of fusing these domains to a well-established yeast two-hybrid protein pair and are currently testing different combinations in order to identify the optimal conditions that will allow us to distinguish between true and false positives.
Other publications describing yeast protein interactions that include work from the laboratory are listed here.
Select the links below to learn about how the two-hybrid systems works:
How to clone 6000 ORFs
Primers
Vectors: pOBD2 and pOAD
Yeast strains
Recombination Cloning Protocols
Dual-reporter 2-hybrid assay
Russell Lo (former lab member)
Although the two-hybrid assay has been useful in discovering protein-protein interactions, a major disadvantage of the approach is the large number of false-positives that arise. Technical false positives activate the transcription of the HIS3 reporter gene but do not represent physical interaction of the two proteins (“bait” and “prey”). To address this issue, we have been working on incorporating a second reporter into the existing system. The second reporter relies on the re-association of the firefly luciferase protein, which has been split in two halves and fused to the bait and prey constructs. With a true bait and prey protein-protein interaction, the two halves of luciferase are brought into proximity and should reconstitute enzyme activity. Thus, true physical interactions should be both transcription positive and luminescence positive, whereas false positives would yield only the transcriptional signal.
Initial tests revealed that the two halves of luciferase were capable of self-associating even in the absence of interacting bait and prey proteins. Splitting of the luciferase protein also led to a large decrease in luminescence upon re-association of the two halves, compared to the intact protein. To address these issues, I performed random mutagenesis on the luciferase gene in order to identify mutations that could either boost luminescence or decrease self-association. Mutations from these two classes were then coupled and the resulting constructs are being tested on several known bait/prey protein pairs as well as on yeast prey proteins that are known false positives. Validation of this new system will require a two-hybrid screen of a library to be performed, yielding results that are enriched in true positives.
Initial tests revealed that the two halves of luciferase were capable of self-associating even in the absence of interacting bait and prey proteins. Splitting of the luciferase protein also led to a large decrease in luminescence upon re-association of the two halves, compared to the intact protein. To address these issues, I performed random mutagenesis on the luciferase gene in order to identify mutations that could either boost luminescence or decrease self-association. Mutations from these two classes were then coupled and the resulting constructs are being tested on several known bait/prey protein pairs as well as on yeast prey proteins that are known false positives. Validation of this new system will require a two-hybrid screen of a library to be performed, yielding results that are enriched in true positives.
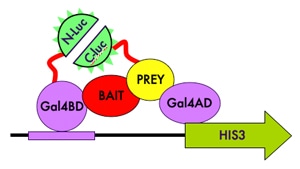
Random addressed protein arrays
Carlos Araya and Doug Fowler (former lab members)
In the last decade, sequencing technologies have seen prodigious improvements in throughput and costs yet, protein activity and enzymatic assays –and hence our ability to query gene function– have not enjoyed concomitant advances. We are interested in applying the high-throughput sequencing technologies to generate an array of clusters containing DNA templates for in vitro transcription/translation (IVTT) reactions. Our goal is to identify each template in the array by sequencing and then generate a high-density protein array by capturing in vitro translated proteins proximal to the template cluster.
Towards this goal we have established methods for clonal amplification of DNA on solid surfaces (solid-phase amplification and emulsion amplification), in vitro transcription and translation methods using home-made cellular extracts, and are working on strategies for capturing proteins on surfaces.
Notably, an array platform containing millions of features will allow an increase in throughput three orders of magnitude above that of current protein arrays. Such increased throughput will enable experiments aimed at mapping protein interactions, generating enzyme activity profiles with activity-based probes and identifying novel enzyme activities to be performed at new scales. The ability to perform massively parallel protein assays should open novel research opportunities in proteomics, metagenomics and directed protein evolution. In addition, such development would also show how high-throughput sequencing platforms can be integrated into new technologies for functional assays, with end-points distinct from sequencing reads.
Towards this goal we have established methods for clonal amplification of DNA on solid surfaces (solid-phase amplification and emulsion amplification), in vitro transcription and translation methods using home-made cellular extracts, and are working on strategies for capturing proteins on surfaces.
Notably, an array platform containing millions of features will allow an increase in throughput three orders of magnitude above that of current protein arrays. Such increased throughput will enable experiments aimed at mapping protein interactions, generating enzyme activity profiles with activity-based probes and identifying novel enzyme activities to be performed at new scales. The ability to perform massively parallel protein assays should open novel research opportunities in proteomics, metagenomics and directed protein evolution. In addition, such development would also show how high-throughput sequencing platforms can be integrated into new technologies for functional assays, with end-points distinct from sequencing reads.
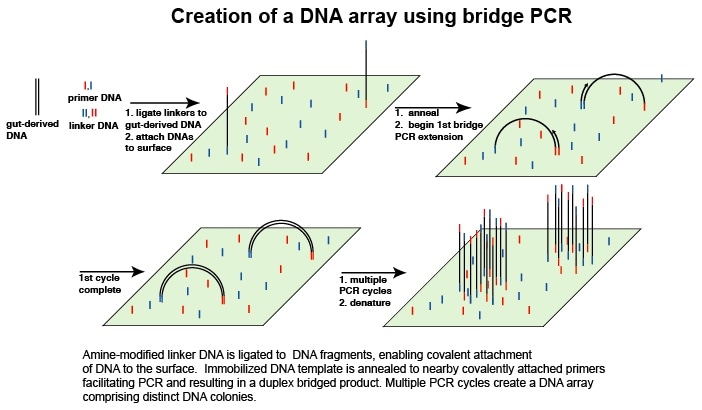
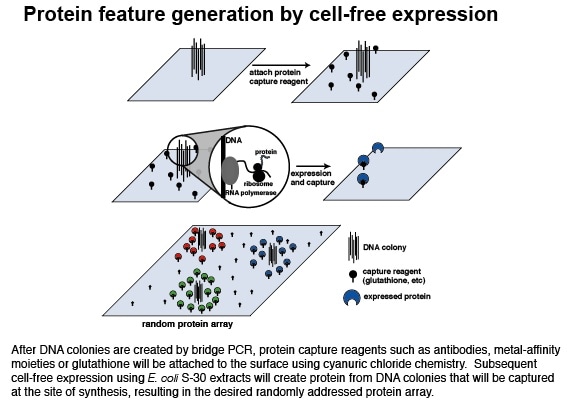
Malaria protein arrays
Anastasia Gridasova (former lab member)
Malaria is one of the world’s most devastating human diseases, affecting an estimated 500 million people and resulting in 2.5 million deaths globally each year. It is caused by four species of the protozoan parasite, Plasmodium, with the most deadly being Plasmodium falciparum. Progress in understanding malaria has been limited by the parasite’s complex life cycle and by difficulties expressing and purifying functional P. falciparum proteins in heterologous systems. With the completion of the P. falciparum genome sequence, a useful tool would be a protein array in which individual parasite proteins are displayed and available for functional assays.
To prepare a P. falciparum protein array, we are using 1000 P. falciparum open reading frames generated by the Structural Genomics of Pathogenic Protozoa (SGPP) consortium. These ORFs will be translated in vitro and printed onto glass slides. Our goal is to use these protein arrays in assays that benefit from rapid, simultaneous and sensitive screening of large numbers of proteins. For example, antigen profiling assays can identify P. falciparum proteins that govern host immune responses against the pathogen. Another application for the array is the biochemical characterization of the “hypothetical” proteins that represent more than 60% of P. falciparum genome, many of which are unique to P. falciparum. To characterize these proteins we will profile arrayed proteins with fluorescent substrate profiling probes.
To prepare a P. falciparum protein array, we are using 1000 P. falciparum open reading frames generated by the Structural Genomics of Pathogenic Protozoa (SGPP) consortium. These ORFs will be translated in vitro and printed onto glass slides. Our goal is to use these protein arrays in assays that benefit from rapid, simultaneous and sensitive screening of large numbers of proteins. For example, antigen profiling assays can identify P. falciparum proteins that govern host immune responses against the pathogen. Another application for the array is the biochemical characterization of the “hypothetical” proteins that represent more than 60% of P. falciparum genome, many of which are unique to P. falciparum. To characterize these proteins we will profile arrayed proteins with fluorescent substrate profiling probes.
Identifying protein targets of ubiquitin ligases
Lea Starita and Russell Lo (former lab members)
Ubiquitin (Ub) is a 76 amino acid protein that when attached to a target protein can alter its fate in multiple ways. Ub is an essential signaling molecule in nearly every pathway in eukaryotic cells. The E1 ubiquitin activating enzyme, E2 ubiquitin conjugating enzymes and E3 ubiquitin ligases act in concert in order to covalently attach a Ub moiety to the epsilon amine on a lysine side chain or less commonly, the free amino group at the N-terminus of a substrate protein via an isopeptide bond. The E3 gives the enzyme cascade substrate specificity. There are 35 E3s in S. cerevisiae and nearly 1000 putative E3s in the human genome. Many proteins important for human disease are E3s, such as the breast cancer-specific tumor suppressor BRCA1 (breast cancer-1) and the early onset-Parkinson’s disease protein Parkin. Therefore, knowing the specific protein substrates of individual E3 enzymes would be of great importance in medicine.
One approach I am taking to determine specific substrates of E3s is as follows:
Individual E3 ubiquitin ligases will be fused to the bacterial biotin ligase, BirA, which attaches biotin to proteins that contain a biotin-acceptor peptide. A second fusion protein will be generated where ubiquitin is fused to a biotin-acceptor peptide fragment. In this arrangement, when the biotin-acceptor/ubiquitin fusion protein is brought to the E3 ubiquitin ligase/BirA fusion protein, the attached BirA will biotinylate the ubiquitin, thus marking it as having been acted on by that specific ubiquitin ligase. Substrate proteins to which the biotinylated ubiquitin is subsequently attached can then be purified using streptavidin chromatography and identified by mass spectrometry. Applying this approach to each individual E3 ubiquitin ligase will provide a critically needed proteome-wide view of ubiquitin ligases and their substrates. Ultimately, I would adapt the system for the study of ubiquitin ligases in mammalian tissue culture cells.
One approach I am taking to determine specific substrates of E3s is as follows:
Individual E3 ubiquitin ligases will be fused to the bacterial biotin ligase, BirA, which attaches biotin to proteins that contain a biotin-acceptor peptide. A second fusion protein will be generated where ubiquitin is fused to a biotin-acceptor peptide fragment. In this arrangement, when the biotin-acceptor/ubiquitin fusion protein is brought to the E3 ubiquitin ligase/BirA fusion protein, the attached BirA will biotinylate the ubiquitin, thus marking it as having been acted on by that specific ubiquitin ligase. Substrate proteins to which the biotinylated ubiquitin is subsequently attached can then be purified using streptavidin chromatography and identified by mass spectrometry. Applying this approach to each individual E3 ubiquitin ligase will provide a critically needed proteome-wide view of ubiquitin ligases and their substrates. Ultimately, I would adapt the system for the study of ubiquitin ligases in mammalian tissue culture cells.
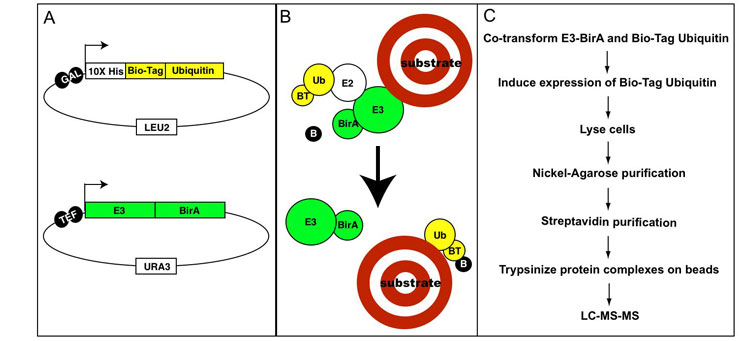
Genomewide identification of spliced introns using a tiling microarray
Zhihong Zhang and Jay Hesselberth (former lab members)
One hallmark of eukaryotic gene structure is the presence of introns, which are spliced out of pre-mRNAs prior to translation. Introns excised from pre-mRNA molecules by the spliceosomal machinery are released in the form of lariats, in which the 5’ end of the intron RNA is linked via a phosphodiester bond to the 2’ hydroxyl of an internal adenosine residue. The lariat must be debranched by 2’-5’ phosphodiesterase prior to their turnover. In the absence or knockdown of the debranching enzyme, these lariat RNAs accumulate. We have carried out a genomewide identification of spliced intron using a genomic tiling microarray in Saccharomyces cerevisiae by comparison of total RNA between DBR+ and dbr1 strains. This approach identified 141 of 272 known introns, confirmed three previously predicted introns, predicted four novel introns (of which two were experimentally confirmed), and led to the reannotation of four others.
DBR homologs and DBR-mediated lariat degradation were also found in other organisms. Currently, we are working on adapting the tiling array approach for genome-wide identification of introns in Drosophila and human cells. It has been reported that knockdown of the debranching enzyme in Drosophila via RNAi can cause lariat stabilization. We also applied this approach to human cell cultures and observed a similar but modest effect. We are now testing different RNAi knockdown approaches in both organisms to improve the efficiency of lariat accumulation. Analysis of lariat accumulation in these complex organisms will not only contribute to their genome annotation, but also extend our understanding of regulated and alternative splicing in these species.
Published Results
Zhang Z, Hesselberth JR, Fields S. Genome-wide identification of spliced introns using a tiling microarray. Genome Res. 2007 Mar 9.
Download PDF
Supplemental Data
DBR homologs and DBR-mediated lariat degradation were also found in other organisms. Currently, we are working on adapting the tiling array approach for genome-wide identification of introns in Drosophila and human cells. It has been reported that knockdown of the debranching enzyme in Drosophila via RNAi can cause lariat stabilization. We also applied this approach to human cell cultures and observed a similar but modest effect. We are now testing different RNAi knockdown approaches in both organisms to improve the efficiency of lariat accumulation. Analysis of lariat accumulation in these complex organisms will not only contribute to their genome annotation, but also extend our understanding of regulated and alternative splicing in these species.
Published Results
Zhang Z, Hesselberth JR, Fields S. Genome-wide identification of spliced introns using a tiling microarray. Genome Res. 2007 Mar 9.
Download PDF
Supplemental Data
Recombination studies
Clem Stanyon (former lab member)
We are interested in turning the process of recombination to our ends, using it to facilitate gene therapy and genome engineering. To this end, we have developed a system that allows for the selection of yeast in which two overlapping parts of the selectable marker for Kanamycin (KanR) are brought together. Our system features a genomic recipient locus and a plasmid donor construct (Figure 1). Recombination between these elements results in the reconstitution of a functional KanR marker when the elements are united through the mating of two yeast strains, each of which carries one of the two elements. We have made and tested a variety of donor structures (Figure 2), together with different effector proteins that localize to the two DNA elements of the system.
The greatest total efficiency of conversion to the KanR phenotype was obtained with the donor construct having the most homology to the recipient locus. We are therefore using this construct to screen a random yeast genomic GAL4 library to isolate peptides that promote homologous repair. To do this, UAS GAL sequences have been cloned into the middle of the recipient locus and on the flanks of the longest donor construct we currently possess; yeast expressing peptides that promote HR when bound to the donor or recipient DNA will more frequently convert to the KanR phenotype. As genome engineering will most likely feature exogenously created linear donor DNA and may also utilize lesion-targeting endonucleases, we are employing the homing endonuclease I-SceI to introduce dsDNA breaks in both the donor and recipient DNA. While this will produce a background, we will screen serially derived libraries for clones enriched by repeated selection for the ability to promote HR.
The greatest total efficiency of conversion to the KanR phenotype was obtained with the donor construct having the most homology to the recipient locus. We are therefore using this construct to screen a random yeast genomic GAL4 library to isolate peptides that promote homologous repair. To do this, UAS GAL sequences have been cloned into the middle of the recipient locus and on the flanks of the longest donor construct we currently possess; yeast expressing peptides that promote HR when bound to the donor or recipient DNA will more frequently convert to the KanR phenotype. As genome engineering will most likely feature exogenously created linear donor DNA and may also utilize lesion-targeting endonucleases, we are employing the homing endonuclease I-SceI to introduce dsDNA breaks in both the donor and recipient DNA. While this will produce a background, we will screen serially derived libraries for clones enriched by repeated selection for the ability to promote HR.
Yeast aging
Trey Powers and Matt Kaeberlein (former lab members)
The budding yeast Saccharomyces cerevisiae serves as a useful organism for studying factors that determine cellular longevity. The aging of mitotically active cells in higher eukaryotes can be modeled by the replicative life span of yeast mother cells, whereas aging of post-mitotic cells more closely resembles the chronological survival of quiescent yeast during stationary phase (Figure 1). We are interested in using high-throughput technologies to identify and characterize genes that modify both aspects of cellular life span.
Replicative aging
Measurement of yeast replicative life span requires micromanipulation of daughter cells away from mother cells following each mitotic cycle. The time-consuming nature of this assay has precluded large-scale analyses of replicative aging. In collaboration with Dr. Brian Kennedy (Department of Biochemistry, University of Washington), we have developed a method to allow semi-quantitative measurement of replicative life span based on the aging properties of a small number of cells. To date, we have determined the replicative life span phenotypes for approximately 20% (~1000 strains) of the ORF deletion collection. Completion of this analysis, in collaboration with the Kennedy lab, should take approximately 2 years.
Based on our analysis to date, we have already made several important discoveries, including the surprising finding that life span extension by calorie restriction (CR) does not require the NAD-dependent histone deacetylase, Sir2. The Sir2-indepenent nature of CR is demonstrated two ways: first, calorie restriction and overexpression of Sir2 increase life span additively, and second, CR increases life span to a greater extent in cells lacking Sir2 (and Fob1) than in wild type cells (Figure 2).
Published Results
Kaeberlein, M., Kirkland, K.T., Fields, S. and Kennedy, B.K. (2004) Sir2-independent life span extension by calorie restriction in yeast. PLoS Biology Sep;2(9):E296.
Download PDF
We have also determined that, contrary to a prior model proposed by Lin, Guarente, and colleagues, CR does not increase yeast life span by enhancing respiration. Yeast cells completely lacking mitochondrial DNA either have a normal life span or a dramatically shortened life span. In both cases, however, CR dramatically enhances longevity, demonstrating that respiration is not required for life span extension by CR.
Published Results
Kaeberlein M, Hu D, Kerr EO, Tsuchiya M, Westman EA, Dang N, Fields S, Kennedy BK. Increased Life Span due to Calorie Restriction in Respiratory-Deficient Yeast. PLoS Genet. 2005 Nov 25;1(5):e69.
Download PDF
Of the first 564 single-gene deletion strains examined, 14 show a significant increase in replicative life span relative to the parental strain. This set includes two overlapping ORFs along with several genes that code for proteins with functions related to the nutrient responsive kinases Tor and Sch9. Of particular interest is the finding that three genes involved in ribosome biogenesis (a Tor and Sch9-regulated process) were among our set of long-lived deletion strains: REI1, RPL31A, and RPL6B. Rpl31a and Rpl6b are protein components of the large ribosomal subunit and Rei1 is a protein of unknown function that we have determined plays a role in large subunit biogenesis. This has led us to propose a model whereby CR increases life span by decreasing Tor and Sch9 activity which results in decreased ribosome biogenesis and translation (Figure 3).
Published Results
Kaeberlein M, Powers RW 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005 Nov 18;310(5751):1193-6.
Download PDF
Chronological aging
In order to examine post-mitotic survival in a high-throughput manner, we have developed a method that allows for the simultaneous determination of chronological life span for several thousand yeast strains in a highly quantitative manner (Figure 4). We have used this technology to screen the ORF deletion collection for genes whose deletion affects chronological aging. From this analysis, we have identified several genes (Table 1) implicated in the TOR pathway that extend chronological life span when deleted (Figure 5). The TOR proteins are highly conserved from yeast to humans and promote cellular growth in response to nutrients, especially amino acids. We have found that limitation of amino acids in the media, or pharmacological inhibition of TOR using rapamycin or methionine sulfoximine (MSX) (Figure 6) can extend chronological life span, similar to deletion of TOR pathway components. Additionally, many of these interventions correlate with an increased nuclear accumulation of the stress-responsive transcription factor Msn2, and a resistance to heat and oxidative stresses. We propose a model by which decreased TOR activity up-regulates the activity of stress-response transcription factors (including Msn2) and thus promotes longevity (Figure 7).
Published Results
Powers RW 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006 Jan 15;20(2):174-84.
Download PDF
Replicative aging
Measurement of yeast replicative life span requires micromanipulation of daughter cells away from mother cells following each mitotic cycle. The time-consuming nature of this assay has precluded large-scale analyses of replicative aging. In collaboration with Dr. Brian Kennedy (Department of Biochemistry, University of Washington), we have developed a method to allow semi-quantitative measurement of replicative life span based on the aging properties of a small number of cells. To date, we have determined the replicative life span phenotypes for approximately 20% (~1000 strains) of the ORF deletion collection. Completion of this analysis, in collaboration with the Kennedy lab, should take approximately 2 years.
Based on our analysis to date, we have already made several important discoveries, including the surprising finding that life span extension by calorie restriction (CR) does not require the NAD-dependent histone deacetylase, Sir2. The Sir2-indepenent nature of CR is demonstrated two ways: first, calorie restriction and overexpression of Sir2 increase life span additively, and second, CR increases life span to a greater extent in cells lacking Sir2 (and Fob1) than in wild type cells (Figure 2).
Published Results
Kaeberlein, M., Kirkland, K.T., Fields, S. and Kennedy, B.K. (2004) Sir2-independent life span extension by calorie restriction in yeast. PLoS Biology Sep;2(9):E296.
Download PDF
We have also determined that, contrary to a prior model proposed by Lin, Guarente, and colleagues, CR does not increase yeast life span by enhancing respiration. Yeast cells completely lacking mitochondrial DNA either have a normal life span or a dramatically shortened life span. In both cases, however, CR dramatically enhances longevity, demonstrating that respiration is not required for life span extension by CR.
Published Results
Kaeberlein M, Hu D, Kerr EO, Tsuchiya M, Westman EA, Dang N, Fields S, Kennedy BK. Increased Life Span due to Calorie Restriction in Respiratory-Deficient Yeast. PLoS Genet. 2005 Nov 25;1(5):e69.
Download PDF
Of the first 564 single-gene deletion strains examined, 14 show a significant increase in replicative life span relative to the parental strain. This set includes two overlapping ORFs along with several genes that code for proteins with functions related to the nutrient responsive kinases Tor and Sch9. Of particular interest is the finding that three genes involved in ribosome biogenesis (a Tor and Sch9-regulated process) were among our set of long-lived deletion strains: REI1, RPL31A, and RPL6B. Rpl31a and Rpl6b are protein components of the large ribosomal subunit and Rei1 is a protein of unknown function that we have determined plays a role in large subunit biogenesis. This has led us to propose a model whereby CR increases life span by decreasing Tor and Sch9 activity which results in decreased ribosome biogenesis and translation (Figure 3).
Published Results
Kaeberlein M, Powers RW 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005 Nov 18;310(5751):1193-6.
Download PDF
Chronological aging
In order to examine post-mitotic survival in a high-throughput manner, we have developed a method that allows for the simultaneous determination of chronological life span for several thousand yeast strains in a highly quantitative manner (Figure 4). We have used this technology to screen the ORF deletion collection for genes whose deletion affects chronological aging. From this analysis, we have identified several genes (Table 1) implicated in the TOR pathway that extend chronological life span when deleted (Figure 5). The TOR proteins are highly conserved from yeast to humans and promote cellular growth in response to nutrients, especially amino acids. We have found that limitation of amino acids in the media, or pharmacological inhibition of TOR using rapamycin or methionine sulfoximine (MSX) (Figure 6) can extend chronological life span, similar to deletion of TOR pathway components. Additionally, many of these interventions correlate with an increased nuclear accumulation of the stress-responsive transcription factor Msn2, and a resistance to heat and oxidative stresses. We propose a model by which decreased TOR activity up-regulates the activity of stress-response transcription factors (including Msn2) and thus promotes longevity (Figure 7).
Published Results
Powers RW 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006 Jan 15;20(2):174-84.
Download PDF
Plasmodium protein-protein interaction project
Doug LaCount and Marissa Vignali (former lab members)
Plasmodium falciparum is a mosquito-borne protozoan parasite responsible for the most severe form of malaria. Over 500 million people worldwide are afflicted with malaria, and each year more than one million people- most of them children - die from these infections. Despite the importance of malaria in global health, much remains to be discovered about the molecular biology of these pathogens. Of the ~5,300 proteins predicted from the P. falciparum genome sequence 60% are classified as hypothetical; this designation means that they have never been studied in Plasmodium and do not have sufficient similarity to characterized proteins in other organisms to allow functional assignments to be made. To begin to understand the functions of these novel proteins, we have identified a large number of protein-protein interactions using high-throughput yeast two-hybrid searches with protein fragments derived from genes expressed in the intraerythrocytic stages of P. falciparum. In collaboration with Prolexys Pharmaceuticals, we performed over 32,000 searches and we identified 2,846 interactions involving 1,308 proteins, which corresponds to approximately a quarter of the proteins predicted from the P. falciparum genome. We identified clusters of interacting proteins likely involved in important processes for the survival and infectivity of the parasite, such as gene regulation and host cell invasion. A large fraction of our interactions involve uncharacterized proteins and thus could lead to a new understanding of the functions of those proteins.
In addition to this, we have performed 10,000 searches with P. falciparum baits against human activation domain libraries, and 11,000 searches with P. vivax baits against P. vivax activation domain libraries. We are currently analyzing these datasets.
This work was funded in part by the Structural Genomics of Pathogenic Protozoans effort led by Wim Hol in the Department of Biochemistry.
Published Results
Lacount DJ, Vignali M, Chettier R, Phansalkar A, Bell R, Hesselberth JR, Schoenfeld LW, Ota I, Sahasrabudhe S, Kurschner C, Fields S, Hughes RE. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature. 2005 Nov 3;438(7064):103-7.
In addition to this, we have performed 10,000 searches with P. falciparum baits against human activation domain libraries, and 11,000 searches with P. vivax baits against P. vivax activation domain libraries. We are currently analyzing these datasets.
This work was funded in part by the Structural Genomics of Pathogenic Protozoans effort led by Wim Hol in the Department of Biochemistry.
Published Results
Lacount DJ, Vignali M, Chettier R, Phansalkar A, Bell R, Hesselberth JR, Schoenfeld LW, Ota I, Sahasrabudhe S, Kurschner C, Fields S, Hughes RE. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature. 2005 Nov 3;438(7064):103-7.
A network of WW domain interactions constructed using protein microarrays
Jay Hesselberth and John Miller (former lab members)
We used protein microarray technology to generate a protein interaction map for twelve of the thirteen WW domains present in proteins of the yeast Saccharomyces cerevisiae (Example figure). We observed a total of 1,158 interactions with these 12 domains, most of which have not previously been described. We analyzed the representation of functional annotations within the network, identifying enrichments for proteins with vacuolar and peroxisomal localization, as well as proteins involved in cofactor biosynthesis and protein turnover. The conservation of primary sequence motifs known to be recognized by WW domains was analyzed in the context of the network, and a comparative genomics approach used to dissect the occurrence of such motifs within the dataset. We analyzed the PY (Pro-Pro-Xaa-Tyr) motif in detail, and propose a novel consensus for the motif based on its conservation among orthologs of the interacting protein. The comparative approach revealed that one of the WW domain-containing proteins has an evolutionarily conserved PY motif, possibly indicating a role for WW domain multimerization in the propagation of signals derived from WW domain binding events.
Published Results
Hesselberth JR, Miller JP, Golob A, Stajich JE, Michaud GA, Fields S. Comparative analysis of Saccharomyces cerevisiae WW domains and their interacting proteins. Genome Biol. 2006 Apr 10;7(4):R30.
Download PDF
Published Results
Hesselberth JR, Miller JP, Golob A, Stajich JE, Michaud GA, Fields S. Comparative analysis of Saccharomyces cerevisiae WW domains and their interacting proteins. Genome Biol. 2006 Apr 10;7(4):R30.
Download PDF
Yeast membrane protein array
John Miller and Russell Lo (former lab members)
We have applied the split-ubiquitin system originally described by Johnsson and Varshavsky (1994) to investigate interactions between integral membrane proteins. In brief, one protein is fused to the N-terminal half of ubiquitin (N-Ub) and a second protein is fused to the C-terminal half of ubiquitin (C-Ub). If the membrane proteins exist in close proximity, they bring the two halves of ubiquitin back together, and endogenous ubiquitin C-terminal hydrolases recognize this “reconstituted” ubiquitin and cleave the peptide bond following the last amino acid residue of ubiquitin. In the modified system of Stagljar et al (1998) there is a transcription factor fused to this residue and, upon cleavage, the transcription factor is released from the membrane to enter the nucleus to activate reporter genes.
We have generated a collection of 705 yeast proteins that are annotated as being in an “integral membrane” environment (643 proteins) along with proteins having amino acid homology to these (62). These proteins were made both as fusions with N-Ub, and as fusions to C-Ub with the transcription factor (C-UbPLV).
We tested our transformants for the successful insertion of an in-frame ORF into our C-UbPLV by screening them for an interaction with a generic “positive control”, a N-UbI fusion protein. Of the 705 proteins generated as C-UbPLV fusions, 365 showed an interaction with this wild-type version, N-UbI, which does not require a protein-protein interaction to bind to C-Ub. This result suggests that the 365 fusions bear an insert; the insert is in frame with the C-UbPLV moiety; there are no nonsense mutations in the insert; and the fusion protein is oriented such that the COOH-terminus bearing the fusion with C-UbPLV is exposed to the cytoplasm.
These 365 integral membrane proteins were screened for interactions against the full set of 705 proteins fused to N-UbG, a mutant form of the N-Ub with an isoleucine to glycine mutation at position 13 (Johnsson and Varshavsky (1994)). The pair-wise interactions between these 365 proteins and the 705 N-Ub fusion proteins were assayed similar to the two-hybrid study of Uetz et al. (2000). A set of 1985 putative interactions between 463 NubG fusions and 270 of the C-UbPLV fusions was found. This number of interactions per protein (~8 on average) is likely high due to the false-positive rate associated with this assay (the magnitude of which is not known). These false-positives are likely to result from the high effective concentration of integral membrane proteins due to both sequestration to a two-dimensional lipid bilayer, and co-transport of membrane proteins along the stages of the secretory pathway; additionally, over-expression of the fusion proteins from an episomal plasmid with an ectopic promoter will likely promote some non-physiological interactions.
An advantage of the membrane-based yeast two-hybrid system is that interactions between proteins can be detected at the physiological site of the interaction. This allows the observation of interactions occurring between proteins of most if not all subcellular membranous compartments (Figure 1). However, we also observe interactions between proteins whose native localization is to distinct compartments, and the system by itself is not informative in regards to the location within the cell of the interaction. In some instances (e.g., potentially the Mst27 and Tna1 interaction in the figure) the interaction may occur in early compartments of the secretory pathway, despite the mature proteins involved having disparate ultimate destinations. In other cases the observed interaction is likely to be non-physiological, and results from mislocalization of one or both of the proteins to an inappropriate compartment due to the fusion moiety.
In order to characterize our dataset with the goal of isolating those interactions that are more probably true-positives, we collaborated with Asa Ben-Hur in William Stafford Noble's group. The approach was to use a learning algorithm, the support vector machine (SVM), to classify the interactions based on the statistics of the assay as well as other datasets from the literature (e.g., synthetic-lethality, localization studies, Gene Ontology annotations, etc.). The SVM was trained using interactions found in this study that are also identified by independent experiments as examples of "true-positives". In addition to these 34 interactions, we included 22 that are supported by one computational approach (Deane et al., 2002), and 7 by a different computational analysis (Jansen et al., 2003). These 63 interactions constitute our highest confidence interactions, and are used by the SVM to identify features of the remaining interactions that support their classification as true interactions.
An interesting outcome from multiple SVM analyses is that 138 interactions are always classified as true positives by the algorithm, and 939 are never classified as true (Figure 2). We will therefore examine the interactions consistently predicted to be true as well as the intermediate interactions to identify potential physiological interactions. A comparison of the features of the 138 interactions that are always classified as true with those of the 138 "worst" interactions that are never classified as true shows that the SVM selects, for most features, the values that would be expected to indicate more physiological interactions as shown in the heat map (Figure 3).
Published Results
Miller J.P., Lo R.S., Ben-Hur A, Desmarais C, Stagljar I, Stafford Noble W, Fields S. Large-scale identification of yeast integral membrane protein interactions. Proc Natl Acad Sci U S A. 2005 Aug 23;102(34):12123-12128.
Download PDF
Supplemental Data
We have generated a collection of 705 yeast proteins that are annotated as being in an “integral membrane” environment (643 proteins) along with proteins having amino acid homology to these (62). These proteins were made both as fusions with N-Ub, and as fusions to C-Ub with the transcription factor (C-UbPLV).
We tested our transformants for the successful insertion of an in-frame ORF into our C-UbPLV by screening them for an interaction with a generic “positive control”, a N-UbI fusion protein. Of the 705 proteins generated as C-UbPLV fusions, 365 showed an interaction with this wild-type version, N-UbI, which does not require a protein-protein interaction to bind to C-Ub. This result suggests that the 365 fusions bear an insert; the insert is in frame with the C-UbPLV moiety; there are no nonsense mutations in the insert; and the fusion protein is oriented such that the COOH-terminus bearing the fusion with C-UbPLV is exposed to the cytoplasm.
These 365 integral membrane proteins were screened for interactions against the full set of 705 proteins fused to N-UbG, a mutant form of the N-Ub with an isoleucine to glycine mutation at position 13 (Johnsson and Varshavsky (1994)). The pair-wise interactions between these 365 proteins and the 705 N-Ub fusion proteins were assayed similar to the two-hybrid study of Uetz et al. (2000). A set of 1985 putative interactions between 463 NubG fusions and 270 of the C-UbPLV fusions was found. This number of interactions per protein (~8 on average) is likely high due to the false-positive rate associated with this assay (the magnitude of which is not known). These false-positives are likely to result from the high effective concentration of integral membrane proteins due to both sequestration to a two-dimensional lipid bilayer, and co-transport of membrane proteins along the stages of the secretory pathway; additionally, over-expression of the fusion proteins from an episomal plasmid with an ectopic promoter will likely promote some non-physiological interactions.
An advantage of the membrane-based yeast two-hybrid system is that interactions between proteins can be detected at the physiological site of the interaction. This allows the observation of interactions occurring between proteins of most if not all subcellular membranous compartments (Figure 1). However, we also observe interactions between proteins whose native localization is to distinct compartments, and the system by itself is not informative in regards to the location within the cell of the interaction. In some instances (e.g., potentially the Mst27 and Tna1 interaction in the figure) the interaction may occur in early compartments of the secretory pathway, despite the mature proteins involved having disparate ultimate destinations. In other cases the observed interaction is likely to be non-physiological, and results from mislocalization of one or both of the proteins to an inappropriate compartment due to the fusion moiety.
In order to characterize our dataset with the goal of isolating those interactions that are more probably true-positives, we collaborated with Asa Ben-Hur in William Stafford Noble's group. The approach was to use a learning algorithm, the support vector machine (SVM), to classify the interactions based on the statistics of the assay as well as other datasets from the literature (e.g., synthetic-lethality, localization studies, Gene Ontology annotations, etc.). The SVM was trained using interactions found in this study that are also identified by independent experiments as examples of "true-positives". In addition to these 34 interactions, we included 22 that are supported by one computational approach (Deane et al., 2002), and 7 by a different computational analysis (Jansen et al., 2003). These 63 interactions constitute our highest confidence interactions, and are used by the SVM to identify features of the remaining interactions that support their classification as true interactions.
An interesting outcome from multiple SVM analyses is that 138 interactions are always classified as true positives by the algorithm, and 939 are never classified as true (Figure 2). We will therefore examine the interactions consistently predicted to be true as well as the intermediate interactions to identify potential physiological interactions. A comparison of the features of the 138 interactions that are always classified as true with those of the 138 "worst" interactions that are never classified as true shows that the SVM selects, for most features, the values that would be expected to indicate more physiological interactions as shown in the heat map (Figure 3).
Published Results
Miller J.P., Lo R.S., Ben-Hur A, Desmarais C, Stagljar I, Stafford Noble W, Fields S. Large-scale identification of yeast integral membrane protein interactions. Proc Natl Acad Sci U S A. 2005 Aug 23;102(34):12123-12128.
Download PDF
Supplemental Data
Specific interests of the yeast 2-hybrid array screening
Tony Hazbun (former lab member)
We were interested in using functional genomics tools to decipher two key biological processes that are dysregulated in cancer – chromosome segregation and chromatin modifications. The focusing of genome-wide tools on specific biological processes has several benefits including: 1) bringing an unbiased approach to investigate the process; 2) discovering new players involved in a process; and 3) providing information to model the process. A further aspect of using focused functional genomics is we can come much closer to extracting information that is saturating for a particular method.
The core tool we used is the genome-wide two-hybrid array technology that has been successfully used in various projects in the lab and in collaboration with other labs. The projects that we have outlined below were of high interest.
Comprehensive two-hybrid interaction map of spindle-associated proteins
The protein interaction information for kinetochore and associated spindle proteins is extensive but far from complete. Using the two-hybrid system, we started the comprehensive analysis of protein-protein interactions involving kinetochore and spindle proteins in collaboration with the Drubin and Barnes labs (UC Berkeley). These interactions are uncovering novel connections within the kinetochore and other cellular pathways.
Chromatin modification dependent protein interaction map
Chromatin is subject to a variety of modifications and the specificity these modifications impart upon protein binding is still poorly understood. In collaboration with Min-Hao Kuo (Michigan State), we were working towards charting protein-protein interactions that are dependent on chromatin modifications. The combination of high-throughput techniques such as the tethered catalysis two-hybrid system and selective experiments will help elucidate the function of the various types of chromatin modifications.
Published Results
Guo, D., Hazbun, T.R., Xu, X.-J., Ng, S.-L., Fields, S. and Kuo, M.-H. (2004) A tethered catalysis two-hybrid system to identify protein-protein interactions requiring post-translational modifications. Nature Biotechnology Jul;22(7):888-892.
Download PDF
Yeast unknown ORF project
In a collaborative effort, the two-hybrid array technology was used in conjunction with groups affiliated with the YRC to decipher the roles of 100 essential and uncharacterized yeast genes (Hazbun et al., 2003). The integration of two-hybrid data with data from additional protein-based technologies such as co-purification and mass spectrometry, localization, and protein structure prediction enabled the functional annotation of a large fraction of these yeast genes. The parallel analysis of genes by four complementary technologies has also enhanced our understanding of the properties of the two-hybrid genome-wide array. The overlap of protein-protein interactions identified by mass spectrometry compared with two-hybrid was very low, although they both predicted similar cellular roles that agreed with localization data and protein structure prediction. The two-hybrid interactions tended to occur between proteins that were annotated in more broadly related biological processes, resulting in Go term assignments (Table 1) that were lower in resolution than the mass spectrometry-based terms. This possibly reflects the tendency of two-hybrid to identify interactions with proteins in related biological processes that are not necessarily part of a core complex. For example, protein interactions identified by mass spectrometry for two unknown proteins suggested a role for inter-related complexes (Figure 1) in DNA repair whereas two-hybrid interactions suggested a role in DNA repair as well as links with other biological processes such as chromosome segregation, sumoylation and ubiquitination.
Published Results
Hazbun, T.R., Malmström, L., Anderson, S., Graczyk, B.J., Fox, B., Riffle, M., Sundin, B.A., Aranda, J.D., McDonald, W.H., Chun, C., Snydsman, B.E., Bradley, P., Muller, E.G.D., Fields, S., Baker, D., Yates, J.R. III and Davis, T.N. (2003) Assigning function to yeast proteins by integration of technologies. Molecular Cell 12:1353-1365.
Download PDF
The core tool we used is the genome-wide two-hybrid array technology that has been successfully used in various projects in the lab and in collaboration with other labs. The projects that we have outlined below were of high interest.
Comprehensive two-hybrid interaction map of spindle-associated proteins
The protein interaction information for kinetochore and associated spindle proteins is extensive but far from complete. Using the two-hybrid system, we started the comprehensive analysis of protein-protein interactions involving kinetochore and spindle proteins in collaboration with the Drubin and Barnes labs (UC Berkeley). These interactions are uncovering novel connections within the kinetochore and other cellular pathways.
Chromatin modification dependent protein interaction map
Chromatin is subject to a variety of modifications and the specificity these modifications impart upon protein binding is still poorly understood. In collaboration with Min-Hao Kuo (Michigan State), we were working towards charting protein-protein interactions that are dependent on chromatin modifications. The combination of high-throughput techniques such as the tethered catalysis two-hybrid system and selective experiments will help elucidate the function of the various types of chromatin modifications.
Published Results
Guo, D., Hazbun, T.R., Xu, X.-J., Ng, S.-L., Fields, S. and Kuo, M.-H. (2004) A tethered catalysis two-hybrid system to identify protein-protein interactions requiring post-translational modifications. Nature Biotechnology Jul;22(7):888-892.
Download PDF
Yeast unknown ORF project
In a collaborative effort, the two-hybrid array technology was used in conjunction with groups affiliated with the YRC to decipher the roles of 100 essential and uncharacterized yeast genes (Hazbun et al., 2003). The integration of two-hybrid data with data from additional protein-based technologies such as co-purification and mass spectrometry, localization, and protein structure prediction enabled the functional annotation of a large fraction of these yeast genes. The parallel analysis of genes by four complementary technologies has also enhanced our understanding of the properties of the two-hybrid genome-wide array. The overlap of protein-protein interactions identified by mass spectrometry compared with two-hybrid was very low, although they both predicted similar cellular roles that agreed with localization data and protein structure prediction. The two-hybrid interactions tended to occur between proteins that were annotated in more broadly related biological processes, resulting in Go term assignments (Table 1) that were lower in resolution than the mass spectrometry-based terms. This possibly reflects the tendency of two-hybrid to identify interactions with proteins in related biological processes that are not necessarily part of a core complex. For example, protein interactions identified by mass spectrometry for two unknown proteins suggested a role for inter-related complexes (Figure 1) in DNA repair whereas two-hybrid interactions suggested a role in DNA repair as well as links with other biological processes such as chromosome segregation, sumoylation and ubiquitination.
Published Results
Hazbun, T.R., Malmström, L., Anderson, S., Graczyk, B.J., Fox, B., Riffle, M., Sundin, B.A., Aranda, J.D., McDonald, W.H., Chun, C., Snydsman, B.E., Bradley, P., Muller, E.G.D., Fields, S., Baker, D., Yates, J.R. III and Davis, T.N. (2003) Assigning function to yeast proteins by integration of technologies. Molecular Cell 12:1353-1365.
Download PDF
A yeast screen for P. falciparum mefloquine resistance genes
Mara Jeffress (former lab member)
Mefloquine is an effective antimalarial drug. Unfortunately, resistant strains of Plasmodium falciparum are beginning to arise. The P. falciparum multi-drug resistant gene (Pfmdr1) encodes an ABC transporter that is often altered in mefloquine resistant strains and is presumed to act as a drug efflux pump. Little else is known about the parasite's mefloquine resistance mechanisms. However, mefloquine-resistant strains have been reported that contain no Pfmdr1 alterations, suggesting that additional genes are involved in mefloquine resistance. As the yeast Saccharomyces cerevisiae is sensitive to mefloquine, I have used it to screen for P. falciparum genes that can confer increased mefloquine resistance. Yeast was transformed with a P. falciparum cDNA library under the control of an S. cerevisiae galactose-inducible promoter, followed by selection on mefloquine. Several mefloquine resistance candidate genes were isolated in this screen. The four with the strongest phenotype were chosen for further analysis. These encode an uncharacterized multi-transmembrane-spanning protein, two small uncharacterized proteins, and a putative Rab GTPase activator. Each was analyzed for degree of mefloquine resistance and multidrug resistance. In addition, the mefloquine resistant P. falciparum strain W2-Mef and its sensitive parent W2 were analyzed by semi-quantitative RT-PCR to determine if any of these candidate genes is upregulated in the resistant strain. One candidate was thus regulated and it has been cloned for expression and drug testing in P. falciparum.
Published Results
Jeffress M, Fields S. (2005) Identification of putative Plasmodium falciparum mefloquine resistance genes. Mol Biochem Parasitol. Feb;139(2):133-9.
Download PDF
Published Results
Jeffress M, Fields S. (2005) Identification of putative Plasmodium falciparum mefloquine resistance genes. Mol Biochem Parasitol. Feb;139(2):133-9.
Download PDF
Interactions of human Toll-like receptors
Victoria Brown-Kennerly and Rachel Brown (former lab members)
Toll-like receptor ‘sensor’ proteins are expressed in epithelia and antigen presenting cells. They are localized to the endoplasmic reticulum, the plasma membrane, and phagosome-lysosome membranes. The family of 10 human receptors, named TLR1 through TLR10, detects various microbial antigens or endogenous 'danger' signals, and subsequently triggers an ancient, highly conserved, innate immune response. TLRs are activated by ligand-induced oligomerization that recruits cytoplasmic signaling molecules to the receptors’ intracellular domains. Among the recruits are the MyD88 protein, and other adapters (TIRAP/Mal, TRIF/TICAM1, TRAM and SARM) that preferentially associate with certain activated receptors to impart some level of signaling specificity. All TLR cytoplasmic domains, as well as all identified signal adapters, harbor a canonical TIR (toll - interleukin - response) domain that mediates protein-protein interactions. All ten TLRs, as well as the tumor necrosis factor receptor and some interleukin receptors, activate the NFkB transcription factor. The proteins that impart specificity to the Toll signal transduction pathways are not fully delineated (Figure 1).
Experiments in mammalian cells usually measure TLR activation by expression of an NFkB -driven reporter gene. However, any given cell may express many different receptors that use MyD88 as an adapter to activate NFkB, and of course other pathways, unrelated to the TLRs, can also activate NFkB. In contrast, the simple eukaryotic yeast S. cerevisiae does not possess endogenous TLRs or any recognizable Toll signaling pathway; therefore in yeast we can directly test protein-protein interactions without interference from endogenous proteins or other signaling pathways. We are expressing human TLR TIR domains in yeast, and using the yeast two-hybrid system to study protein interactions in the signal transduction pathway.
We screened for new proteins that bind to the receptors’ cytoplasmic domains, and have found many new interacting proteins that appear to specifically associate with certain TLR cytoplasmic domains. In particular, we have found novel and specific interactions for the closely related group of TLRs 1, 2, 6, & 10 (Table 1). These are candidate proteins that may affect signaling by TLR2 heterocomplexes.
We also performed a structure-function study of the TLR-MyD88 interaction. We mapped the amino acids required for TLR association with this ‘universal’ adapter protein by swapping pieces of TLR2 into the closely related TLRs 1, 6, and 10. This creates chimaeric proteins with new (MyD88-binding) function, and is allowing us to define the exact amino acid differences between MyD88-binding and non-binding TLRs (Figure 2).
The TLR-TIR domains are homologous to each other but not identical; therefore, examining protein interactions with these domains will lend insight into interaction specificity and how structure relates to function for the human TLRs.
Published Results
Brown V, Brown RA, Ozinsky A, Hesselberth JR, Fields S. Binding specificity of Toll-like receptor cytoplasmic domains. Eur J Immunol. 2006 Mar;36(3):742-53.
Download PDF
Experiments in mammalian cells usually measure TLR activation by expression of an NFkB -driven reporter gene. However, any given cell may express many different receptors that use MyD88 as an adapter to activate NFkB, and of course other pathways, unrelated to the TLRs, can also activate NFkB. In contrast, the simple eukaryotic yeast S. cerevisiae does not possess endogenous TLRs or any recognizable Toll signaling pathway; therefore in yeast we can directly test protein-protein interactions without interference from endogenous proteins or other signaling pathways. We are expressing human TLR TIR domains in yeast, and using the yeast two-hybrid system to study protein interactions in the signal transduction pathway.
We screened for new proteins that bind to the receptors’ cytoplasmic domains, and have found many new interacting proteins that appear to specifically associate with certain TLR cytoplasmic domains. In particular, we have found novel and specific interactions for the closely related group of TLRs 1, 2, 6, & 10 (Table 1). These are candidate proteins that may affect signaling by TLR2 heterocomplexes.
We also performed a structure-function study of the TLR-MyD88 interaction. We mapped the amino acids required for TLR association with this ‘universal’ adapter protein by swapping pieces of TLR2 into the closely related TLRs 1, 6, and 10. This creates chimaeric proteins with new (MyD88-binding) function, and is allowing us to define the exact amino acid differences between MyD88-binding and non-binding TLRs (Figure 2).
The TLR-TIR domains are homologous to each other but not identical; therefore, examining protein interactions with these domains will lend insight into interaction specificity and how structure relates to function for the human TLRs.
Published Results
Brown V, Brown RA, Ozinsky A, Hesselberth JR, Fields S. Binding specificity of Toll-like receptor cytoplasmic domains. Eur J Immunol. 2006 Mar;36(3):742-53.
Download PDF
Genetic screening using leucine zippers
Mike DeVit and Meg Branson (former lab members)
We developed a novel type of genetic screening method to identify proteins that function in a common pathway or process. The screen takes advantage of the observation that cellular processes are often initiated when a signal or upstream event causes two or more proteins to physically interact. These are usually part of a cascade of interactions that ultimately lead to the activation of the cellular process. We tested the idea that it might be possible to artificially force these interactions to occur and activate a process in the absence of its normal signal (i.e., cause a gain-of-function phenotype).
We were artificially forcing proteins to interact by fusing them to the leucine zippers from the mammalian Fos and Jun proteins. Fos and Jun leucine zippers form a stable heterodimer that can act as a tether to bring the attached proteins into close physical proximity. Using GFP and proteins that occur in specific subcellular localizations, we have shown that Fos and Jun can cause proteins to co-localize in yeast.
If artificial tethering mimics normal protein-protein interactions and recreates an activity that normally requires an upstream signal or event, it can then be used as a method to genetically screen for unknown members of a process. We can tether every yeast protein to a known component of the pathway (i.e. by coexpressing the known protein as a fusion with the Jun leucine zipper and a library of all yeast proteins fused to Fos) and look for a phenotype associated with activation of the process under study. The process should only be activated when the normal components are tethered to the known one. Since these will be fused to the Fos leucine zipper on a plasmid they can be easily identified.
A collection of all yeast proteins fused to the Fos leucine zipper might also be a useful reagent for tagging proteins. The tag can be added simply by introducing the tag (GFP, protein A, or any other polypeptide tag) fused to the Jun leucine zipper into yeast also expressing Fos fusions.
Published Results
Devit M, Cullen PJ, Branson M, Sprague GF Jr, Fields S. (2005) Forcing interactions as a genetic screen to identify proteins that exert a defined activity. Genome Res. Apr;15(4):560-5.
Download PDF
We were artificially forcing proteins to interact by fusing them to the leucine zippers from the mammalian Fos and Jun proteins. Fos and Jun leucine zippers form a stable heterodimer that can act as a tether to bring the attached proteins into close physical proximity. Using GFP and proteins that occur in specific subcellular localizations, we have shown that Fos and Jun can cause proteins to co-localize in yeast.
If artificial tethering mimics normal protein-protein interactions and recreates an activity that normally requires an upstream signal or event, it can then be used as a method to genetically screen for unknown members of a process. We can tether every yeast protein to a known component of the pathway (i.e. by coexpressing the known protein as a fusion with the Jun leucine zipper and a library of all yeast proteins fused to Fos) and look for a phenotype associated with activation of the process under study. The process should only be activated when the normal components are tethered to the known one. Since these will be fused to the Fos leucine zipper on a plasmid they can be easily identified.
A collection of all yeast proteins fused to the Fos leucine zipper might also be a useful reagent for tagging proteins. The tag can be added simply by introducing the tag (GFP, protein A, or any other polypeptide tag) fused to the Jun leucine zipper into yeast also expressing Fos fusions.
Published Results
Devit M, Cullen PJ, Branson M, Sprague GF Jr, Fields S. (2005) Forcing interactions as a genetic screen to identify proteins that exert a defined activity. Genome Res. Apr;15(4):560-5.
Download PDF
Chemical Profiling of the Yeast Deletion Collection
Chandra Tucker (former lab member)
Understanding the actions of drugs and toxins in a cell is of critical importance to medicine, yet many of the molecular events involved in chemical resistance are relatively uncharacterized. In order to identify the cellular processes and pathways targeted by chemicals, we took advantage of the haploid Saccharomyces cerevisiae deletion strains. Although ~4800 of the strains are viable, the loss of a gene in a pathway affected by a drug can lead to a synthetic lethal effect in which the combination of a deletion and a normally sublethal dose of a chemical results in loss of viability. We carried out genome-wide screens to determine quantitative sensitivities of the deletion set to four chemicals: hydrogen peroxide, menadione, ibuprofen, and mefloquine. Hydrogen peroxide and menadione induce oxidative stress in the cell, whereas ibuprofen and mefloquine are toxic to yeast by unknown mechanisms. Here we report the sensitivities of 659 deletion strains that are sensitive to one or more of these four compounds, including 163 multi-chemical sensitive strains, 394 strains specific to hydrogen peroxide and/or menadione, 47 specific to ibuprofen, and 55 specific to mefloquine. We correlate these results with data from other large-scale studies to yield novel insights into cellular function.
Published Results
Tucker, C.L. and Fields, S. (2004) Quantitative genome-wide analysis of yeast deletion strain sensitivities to oxidative and chemical stress. Comparative and Functional Genomics 5:216-224.
Supplemental Data
Download PDF
Published Results
Tucker, C.L. and Fields, S. (2004) Quantitative genome-wide analysis of yeast deletion strain sensitivities to oxidative and chemical stress. Comparative and Functional Genomics 5:216-224.
Supplemental Data
Download PDF